Genética Enfermedad de Fabry: Desde el Aminoácido a la Clínica
Introducción
La
medicina moderna es el resultado de la sumatoria de esfuerzos en todas las
áreas del conocimiento, en la que todas ellas participan aportando la
información necesaria, que nos permita entender al individuo enfermo en toda su
magnitud, desde lo absolutamente tangible hasta lo intangible; pero creíble
según nuestra orientación personal, por lo que cada día deberíamos estar más
capacitados para conocer al SER HUMANO en su máxima expresión. La clínica no es
más que la resultante de la expresión de todos y cada uno de los procesos
celulares y subcelulares, que actuando de manera sincrónica dan un todo que
puede ser objetivo y cuantificable, por lo que en medida que se conozca más y mejor a cada uno
de los procesos y mecanismos fisiológicos o fisiopatológicos podremos tener más
y mejor información de lo que ocurre en el paciente; es por ello que el estudio
de las Ciencias Básicas cada día debería ocupar mayor espacio en el concierto
del conocimiento médico, para que la Medicina sea cada día más profesional y menos
empírica. Las Enfermedades de Depósito Lisosomal (EDL) constituyen un grupo de patologías en las que es
indispensable el abordaje de las Ciencias Básicas para su comprensión, y las
mismas son un ejemplo que rompe con la dicotomía aún no entendida entre las
Ciencias Básicas y las Ciencias Clínicas, puesto que permite contemporizar
entre ambos mundos, mutuamente incluyentes; es por ello, que con este trabajo
se trata de dar una visión amplia de la Enfermedad de Fabry, haciendo un recorrido por
sus características histomorfológicas, fisiológicas, fisiopatológicas y
terapéuticas, que permitan alcanzar el análisis integral de esta entidad nosográfica.
I.-¿Que son los Lisosomas?
En el citoplasma de las células animales
se encuentran dispersos pequeños sacos de enzimas digestivas denominados
lisosomas, los cuales son muy importantes para el catabolismo y el reciclado de
macromoléculas dentro de una célula. Los lisosomas fueron descubiertos hace más
de 50 años, como resultado de la asociación entre un número de hidrolasas,
siendo una organela con propiedades definidas de centrifugación. Estas
hidrolasas mostraron una latencia relacionada a la estructura, la cual depende
de la integridad de la membrana que las rodea. Desde un inicio se reconoció que
los lisosomas mostraban una morfología heterogénea, conteniendo depósitos
densos y una membrana espiralada. (Fig. No. 1). En las células de mamíferos, se
observan como organelas de ~ 0.5 μm de diámetro, frecuentemente con un
núcleo electrón-denso, los mismos pueden llegar a ocupar entre 0.5 a 5 % del volumen celular
y se concentran cerca de los microtúbulos, que se organizan en el centro de la
célula. Los lisosomas han sido considerados como el compartimiento terminal de
degradación de la vía endocítica y que de acuerdo al tamaño de las partículas
ingeridas, dicha vía puede ser dividida en: pinocitosis (vesículas < 150 nm)
y en fagocitosis (partículas > 250 nm); incluso participan en la autofagia,
crinofagia y proteolisis de algunas proteínas citosólicas transportadas a
través de la membrana lisosomal. La consideración del lisosoma como una simple
"unidad de depósito de basura" ha cambiado recientemente, debido a un mejor
entendimiento de cómo se entrega el material endocitado al lisosoma e incluso
por la identificación de lisosomas que secretan su contenido después de unirse
a la membrana plasmática [1]. Un lisosoma típico contiene más o menos 50
enzimas diferentes que degradan moléculas complejas, incluyendo lípidos,
proteínas, carbohidratos y ácidos nucléicos, que se originan dentro y fuera de
la célula, entre dichas enzimas se encuentran: proteasas, lipasas,
fosfolipasas, glucosidasas, fosfatasas, sulfatasas, nucleasas; estas enzimas son
producidas en el retículo endoplasmático rugoso (RER) y son posteriormente
modificadas en el Aparato de Golgi (AG), en donde se identifican y distribuyen
en los lisosomas (Fig. 2). Las moléculas de bajo peso molecular producto de la
digestión enzimática pueden ser transportados a través de la membrana lisosomal
al citosol. Las enzimas lisosomales exhiben una propiedad importante: todas
tiene actividad óptima a pH ácido (pH=4.6), por lo que son hidrolasas ácidas [1,2].
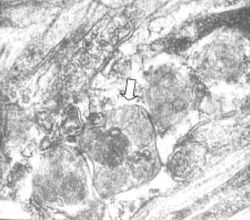
Figura 1.-Se
observan depósitos granulares y laminares en el interior del lisosoma (Flecha)
(Micrografía electrónica 120.000X) Tomado de: de la Sota P y col [3]
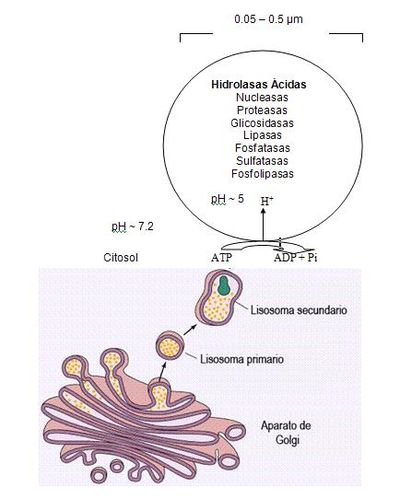
Figura 2.-Esquema de
Lisosoma con sus enzimas y sitio de modificación. Tomado de Luzio y col [1]
La alta concentración interna de protones
se mantienen por un constante transporte de protones (H+-ATPasa)
presente en las membranas de la organela (Fig. 2). La membrana lisosomal
contiene una variedad de proteínas integrales ácidas altamente glicosiladas,
que probablemente la protegen del ataque por parte de las enzimas que la
constituyen. [2]Las enzimas lisosomales son sintetizadas
en el RER, usualmente como formas precursoras que sufren procesos post-
translacionales (fragmentación proteolítico, la adición de oligosacáridos en el
extremo N-terminal y la síntesis de marcadores de reconocimiento, como la
manosa-6-fosfato), previo a formar los lisosomas primarios. Estos lisosomas se
fusionan con otras vesículas, incluyendo fagosomas y autofagosomas que
contienen macromoléculas, detritus celulares y organelas celulares, para formar
lisosomas secundarios donde ocurre la degradación de las moléculas. Mediante la
endocitosis, los lisosomas actúan en la captación de la vitamina B12,
lipoproteínas, hormonas peptídicas y factores de crecimiento [4]. El proceso de síntesis de las enzimas
lisosomales comienza cuando el RNAm se une a un ribosoma libre, comienzan a ser
sintetizadas las enzimas lisosomales. Los polipéptidos ensamblados en los
ribosomas unidos a la membrana, contienen una secuencia señal, de 6 a 12 residuos de aminoácidos
no polares, que llevan al polipéptido naciente a la membrana del RE y permite
la compartamentalización del polipéptido dentro de la luz del RE. Con frecuencia,
el polipéptido señal se ubica cerca del extremo N-terminal. Una vez que sale
del ribosoma, la secuencia señal es reconocida por una "partícula de
reconocimiento de señal" (PRS), la cual está formada por 6 polipéptidos
distintos y una pequeña molécula de RNA, llamada 7SL RNA. El PRS se enlaza
tanto a la secuencia señal del polipéptido naciente como al ribosoma,
impidiendo posteriormente la síntesis del polipéptido y previniendo
plegamientos prematuros de residuos amino-terminales aberrantes.
Se detiene la traslación, deteniendo el enlazamiento del PRS hasta que el
complejo haga contacto y se unan a la membrana del RE. El PRS es una marcador que permite que
todo el complejo (PRS-ribosoma-polipéptido naciente) se una a la superficie
citoplasmática de la membrana del RE. La unión ocurre a través de por lo menos
2 tipos de interacciones: una entre el PRS y el receptor del mismo, y la otra
entre el ribosoma y una proteína de la membrana del canal, llamada translocón. Una vez que el ribosoma se une firmemente a
la membrana del RE, el asa terminal del polipéptido naciente es insertado
dentro de un canal acuoso del translocón; dicha unión parece disparar un cambio
conformacional que abre el canal a la luz del RE. En pasos posteriores,
el PRS es liberado de su receptor en el RE, se reanuda la traslación, y el
péptido es translocado a través del canal, a la luz del RE. Una vez terminada
la translación, el ribosoma unido a la membrana es liberado, se completa el
proceso y se cierra el canal de la membrana. Muchos de los pasos relacionados con la
síntesis de proteínas secretoras y lisosomales, son regulados por la unión o
hidrólisis de GTP. La interacción del PRS con un ribosoma parece inducir
la unión de GTP al PRS, lo cual activa al PRS y le permite acoplarse con el
receptor en la membrana del RE [2] (Fig.3). Las proteínas lisosomales sintetizadas en
los ribosomas unidos a la membrana del RER, entonces son transportadas a la
cisterna Cis del aparato de Golgi junto con otros tipos de proteínas. Una vez
que se encuentran en las cisternas, las enzimas lisosomales solubles son
reconocidas por enzimas que unen un grupo fosfato a la manosa de las cadenas de
carbohidratos unidas al N-terminal. En el cis-Golgi
se fosforilan uno o más residuos de manosa en el oligosacárido Man8(GlcNAc)2
mediante dos reacciones secuenciales.En la primera reacción, se añade un
residuo de N-acetilglucosamina-fosfato al átomo de carbono 6 de la manosa en el
oligosacárido unido al amino terminal por acción de la N-acetilglucosamina
fosfotransferasa, una enzima específica para las enzimas lisosomales. En la
segunda reacción, el residuo de N-acetilglucosamina es removido por una
fosfodiesterasa, dejando un residuo de manosa-6-fosfato (M6P)[5] .
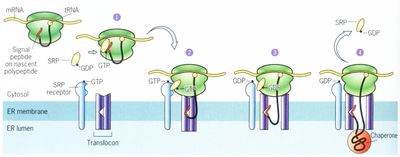
Figura 3.-Esquema de Sistema de Membrana Citoplasmática: Estructura, Funcióny Tráfico en la Membrana. Tomado
de McGovern M. [4].
A diferencia de otras glicoproteínas
distribuidas en la red trans-Golgi (TGN), las enzimas lisosomales poseen
residuos de manosa fosforilados, que actúan como señales de reconocimiento. Las
enzimas lisosomales que contienen esta señal son reconocidas y capturadas por
receptores de manosa-6-fosfato (MPR), que son proteínas integrales de la
membrana, concentradas dentro de una hendidura cubierta de clatrina del TGN .
Se piensa que los receptores de manosa-6-fosfato atraviesan la membrana del TGN
en diferentes sitios de unión, por lugares opuestos de la membrana. Mientras
que una parte de los receptores de la manosa-6-fosfato, se proyectan hacia
dentro de la luz del TGN, este se une a una enzima lisosomal, y una parte del
mismo se proyecta hacia el citosol y se une a un complejo adaptador-clatrina
que se enlaza a la superficie citosólica de la membrana del TGN. Estas
interacciones aseguran que las enzimas lisosomales sean englobadas en vesículas
cubiertas de clatrina, y que una vez liberadas al citoplasma, las mismas se
dirijan a un destino particular que son los endosomas tardíos y posteriormente
darán origen a los lisosomas. Los MPRs se disocian de las enzimas lisosomales
antes de llegar al lisosoma, y los mismos son devueltos al TGN en vesículas sin
cubierta. Los receptores de manosa-6-fosfato están
localizados en la membrana plasmática, donde son capaces de atrapar enzimas
lisosomales que son secretadas al espacio extracelular, y se reintegran a las
enzimas en una vía que las devuelve a los lisosomas.La vía de distribución de la
manosa-6-fosfato para las enzimas lisosomales señala varios principios
importantes que se aplican a la distribución de las proteínas secretoras y de
membrana. Primero, el residuo de manosa-6-fosfato es una de las señales de distribución que llevan a las
proteínas a diferentes compartimientos dentro de la vía secretora. Segundo, los
receptores de membrana difunden con sus ligandos dentro de discretas regiones
de la membrana de una organela, donde son incorporados dentro de vesículas de
transporte. Tercero, estas vesículas de transporte se unen solamente con una
organela específica, el endosoma tardío. Y finalmente, los receptores de
transporte celular, son reciclados después de que se disocian de sus ligandos [2].
NOTA:Toda la información que se brinda en este artículo es de carácter investigativo y con fines académicos y de actualización para estudiantes y profesionales de la salud. En ningún caso es de carácter general ni sustituye el asesoramiento de un médico. Ante cualquier duda que pueda tener sobre su estado de salud, consulte con su médico o especialista.
Instituto de Medicina Tropical - Facultad de Medicina - Universidad Central de Venezuela.
Elaborado por el Centro de Análisis de Imágenes Biomédicas Computarizadas CAIBCO, caibco@ucv.ve
Este portal ha sido desarrollado gracias al apoyo del Fonacit