Genética Enfermedad de Fabry: Desde el Aminoácido a la Clínica
Histología de la Enfermedad de Fabry
Estudios realizados empleando
técnicas de microscopia de luz y electrónica para evaluar los cambios
histológicos en tejidos de ratones knockout para α-Gal-A, mostraron que
las células con mayor alteraciones son los macrófagos y las células derivadas
de macrófagos del hígado y la piel, y las células musculares lisas que rodean a
los vasos sanguíneos y el corazón [14]. En un estudio realizado en un paciente con enfermedad
de Fabry, la mayor concentración de globotriaocilceramida (Gb3) se reportó
primero en los riñones, seguido del corazón y el hígado; y esta fue 35 veces
mayor en el riñón que en el hígado [16]; contrariamente cuando se
hizo la misma observación en ratones knockout Gb3 fue similar en ambos
tejidos. Otro aspecto interesante de resaltar de este estudio con ratones
knockout son los obtenidos con el transplante de médula ósea, en donde en un
número de órganos, la concentración de Gb3 fue significativamente menor en los
ratones transplantados. El transplante alogénico de médula ósea, como fuente de
enzima normal, ha indicado ser beneficioso en estudios humanos con otras
enfermedades lisosomales, pero nunca ha sido utilizado en la enfermedad de
Fabry [14,16]. En ratones knockout tratados con
transplante de médula ósea, la actividad enzimática de α-Gal-A en el
hígado fue de 10% con respecto a los niveles normales, lo que indica que la
acumulación de Gb3 fue completamente depurada de éste órgano. En experimentos
con cultivos de fibroblastos, bajos niveles de la actividad de α-Gal-A
(menor de 5%) fueron capaces de normalizar el metabolismo del sustrato [14].
En las células cardíacas, los depósitos de Gb3 provocan una hipertrofia
progresiva, y no es un fenómeno específico, ya que se observa en otras
enfermedades de depósito lisosomal. Los depósitos observados en las células
endoteliales están asociados con hipertrofia de la membrana basal del capilar y
se han observado con o sin ningún grado leve de depósitos, lesiones ni en los
sinusoides de la corteza de la glándula adrenal ni en el hígado. Los glomérulos
renales muestran hialinización focal que se inicia en la región mesangial y las
células tubulares proximales están libres de depósitos [17]. En los
podocitos y en las células del epitelio tubular distal es donde se han
observado la mayor cantidad de depósitos intracitoplasmáticos. La observación
de los depósitos con el microscopio electrónico muestra inclusiones osmofílicas
típicas en el citoplasma de todos los tipos de células renales y se observa la
típica imagen de "piel de cebolla" o "apariencia de cebra". Estos hallazgos
patológicos son evidentes en pacientes homocigotos o heterocigotos, y ocurren
antes de que se desarrolle la enfermedad renal clínica [18]. Las paredes de las arterias coronarias, renales e
intrarrenales muestran signos de arteriopatía, con un importante compromiso de
las células del músculo liso vascular. La arteriopatía se inicia con los
depósitos, seguidos por la degeneración celular, depósito en la matriz
extracelular y calcificación confinada a la pared vascular. Ocasionalmente, las
células musculares lisas muestran necrosis, con dispersión de depósitos
lipídicos dentro del citoplasma. Se han observado fibroblastos con grado
variable de depósitos a nivel de la capa íntima de los vasos y de la válvula
mitral, y esto está asociado con la presencia de pérdida de células y necrosis.
Igualmente se han observado grandes depósitos a nivel de las células de Leyding
y en epitelio del epidídimo [17]. La enfermedad de Fabry, junto con la
enfermedad de Batten-Kufs´ (lipofuscinosis ceroide) y la manosidosis, forman un
grupo de enfermedades de depósito lisosomal con manifestación asintomática en
el músculo esquelético (Fig. 5), a diferencia de lo que ocurre en la Enfermedad de Pompe,
que es otra enfermedad de depósito lisosomal, por deficiencia de la α-1,4
glucosidasa; pero con compromiso sintomático del músculo esquelético [19,20].
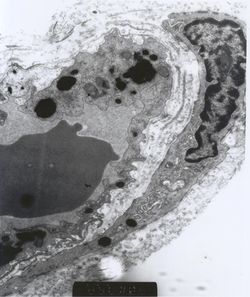
Fig. 5.- Micrografía de un capilar de un músculo
esquelético. Se observa un endotelio irregularmente hipertrófico, con
obstrucción parcial de la luz del capilar. Depósitos electrondensos de Gb3 en
el endotelio y en el pericito. (10.000X).
NOTA:Toda la información que se brinda en este artículo es de carácter investigativo y con fines académicos y de actualización para estudiantes y profesionales de la salud. En ningún caso es de carácter general ni sustituye el asesoramiento de un médico. Ante cualquier duda que pueda tener sobre su estado de salud, consulte con su médico o especialista.
Instituto de Medicina Tropical - Facultad de Medicina - Universidad Central de Venezuela.
Elaborado por el Centro de Análisis de Imágenes Biomédicas Computarizadas CAIBCO, caibco@ucv.ve
Este portal ha sido desarrollado gracias al apoyo del Fonacit