Maitane Barreda
Estudiante IV año Medicina, Escuela de Medicina José María Vargas, Facultad de Medicina, Universidad Central de Venezuela, Caracas, Venezuela
Katherine Lancianese
Estudiante IV año Medicina, Escuela de Medicina José María Vargas, Facultad de Medicina, Universidad Central de Venezuela, Caracas, Venezuela
Cristina Matheus
Estudiante IV año Medicina, Escuela de Medicina José María Vargas, Facultad de Medicina, Universidad Central de Venezuela, Caracas, Venezuela
Michelle Merheb
Estudiante IV año Medicina, Escuela de Medicina José María Vargas, Facultad de Medicina, Universidad Central de Venezuela, Caracas, Venezuela
Cristin Wehbe
Estudiante IV año Medicina, Escuela de Medicina José María Vargas, Facultad de Medicina, Universidad Central de Venezuela, Caracas, Venezuela
Marcel Jesús Marcano-Lozada
marcelmarcano@gmail.com
Docente Asistente Cátedra de Microbiología, Escuela de Medicina José María Vargas, Facultad de Medicina, Universidad Central de Venezuela, Unidad de Microbiología Médica, Angios Centro Vascular y de Cuidado Integral de Heridas, Caracas, Venezuela
Stephanie Barbeito
Estudiante IV año Medicina, Escuela de Medicina José María Vargas, Facultad de Medicina, Universidad Central de Venezuela, Caracas, Venezuela
Microbiología Úlceras crónicas: modelo de integración entre patología vascular, inmunológica e infecciosa Fecha de recepción: 20/06/2011
Fecha de aceptación:
29/08/2011
Se conocen como úlceras crónicas aquellas heridas que no cicatrizan en un período largo de tiempo, caracterizadas por presentar un área necrótica en el sitio de lesión con un centro purulento y sanguinolento, causando dolor en la persona que la padece. Causan una respuesta inflamatoria crónica produciendo un daño mayor en el tejido y evitando la cicatrización del mismo. Planteamos un modelo que relaciona la gravedad de las úlceras vasculares (venosas y/o arteriales), infecciosas (Mycobacterium ulcerans) y las asociadas a alteraciones inmunológicas (desbalance entre el perfil de citocinas proinflamatorias y antiinflamatorias), lo que conlleva a un proceso de inflamación crónica evitando la cicatrización del tejido ulcerado.
Title Chronic ulcers: an integration model of vascular, immunological & infectious pathologies
Abstract Chronic ulcers are those that do not heal during a long period of time. They are characterized by necrosis of the injured tissue with a purulent center and bleeding, being a painful condition. They are caused by an exaggerated inflammatory response producing major damage in the tissue and interfering with cicatrization. This model shows the relationship between the effects caused by vascular problems (venous and/or arterial), those caused by infectious agents (Mycobacterium ulcerans) and the immunological alterations that happen as a consequence of the evolution of the ulcer (unbalanced profile of proinflamatory and anti-inflammatory citocines), leading to a chronic process of inflammation retarding cicatrization of the ulcered tissue.
Key Word chronic ulcers, cytokines, cicatrization, Mycobacterium ulcerans
Úlceras crónicas: modelo de integración entre patología vascular, inmunológica e infecciosa
Introducción
Las úlceras crónicas se caracterizan por la pérdida de
la continuidad del tejido con múltiples características como lesiones cutáneas
con presencia de necrosis alrededor, centro purulento y sangrante, acompañadas
de un proceso inflamatorio y escasa tendencia a la cicatrización del epitelio y
mucosa que se encuentran expuestos a la degeneración por la aparición de la
úlcera, así como erosiones cutáneas con fondo límpido y bordes prominentes que limitan
el área (1).
Las úlceras crónicas se pueden clasificar en úlceras
vasculares (venosas o arteriales), infecciosas e inmunitarias.
Las úlceras vasculares son aquellas que dañan el
endotelio vascular como consecuencia de una patología arterial o venosa
obstructiva. Muestran una incidencia de 2,5 millones de casos registrados
anualmente en los Estados Unidos. Éstas suelen ser recurrentes, causan
incapacidad y limitan la deambulación.(2)
Enfocándonos en las úlceras venosas, aparecen por lo
general en la pierna en la región supramaleolar interna, aunque también se
pueden encontrar en la zona de las pantorrillas representando un 80-90 % total
de todas las úlceras vasculares. Son indoloras y en la periferia suele existir
una vena insuficiente.(3)
La alta incidencia de
población que padece úlceras vasculares, las prolongadas estancias
hospitalarias y las largas bajas laborales, hacen que el tratamiento local de
este tipo de lesiones sea un reto para el personal de salud. La elección del
tratamiento se hará tomando en cuenta ciertos criterios de valoración que
incluyen el estado general del paciente, patología base o el proceso causante
de la lesión, así como también basándose en las características propias de la
úlcera tamaño, profundidad, localización, presencia de tejido necrótico,
infección, valoración de la zona periulcerosa, productos utilizados
anteriormente, entre otros.(2)
Existen también úlceras causadas por desordenes
metabólicos como la Diabetes Miellitus, especialmente en personas que presentan
pie diabético. Donde se pierde el engrosamiento de la pared de los capilares
afectando el transporte transcapilar de las células, aumentando la expresión de
moléculas de adhesión que favorecen la extravasación y por ende el incremento
de células inflamatorias en el sitio de la lesión. Se encuentra una disminución
de las células encargadas de la cicatrización y hay presencia de necrosis del
tejido alrededor de la úlcera. En estas lesiones el componente macro y
microangiopático asociado a la glicosilación por el exceso de glucosa sanguínea
lidera una serie de fenómenos inflamatorios e inmunológicos que conllevan a que
la lesión sea incapaz de sanar.(4)
En los últimos años se ha registrado un aumento de la
incidencia de las úlceras en general, siendo esto muy importante a nivel
mundial debido a que eltratamiento para
cada una de ellas es limitado.
Un tipo de úlcera encontrada con gran incidencia es de
naturaleza infecciosa, donde destacan las infecciones parasitarias, virales y
bacterianas (leishmaniosis, Hansen, herpéticas, sifilíticas), pero el prototipo
de úlcera crónica, herida que no sana es la úlcera de Buruli, causada por la
infección de Mycobacterium ulcerans, única micobacteria conocida hasta
la actualidad con la capacidad de generar una toxina llamada Mycolactona. Se
piensa que la micobacteria genera la úlcera en el músculo esquelético causando
la inhibición de la respuesta inmunitaria en el sitio de lesión
(inmunosupresión local).(1)
Algunos de los tratamientos empleados en la actualidad
para tratar las úlceras de Buruli consiste en antimicrobianos como la
rifampicina y estreptomicina, que se encargan de reinvertir el proceso de
inmunosupresión local y conducir al proceso inflamatorio habitual en diferentes
compartimientos de la piel.(5)
Las úlceras vasculares se pueden tratar con DHN-Na
(difenilhidantoinato de sodio) favoreciendo la cicatrización de las lesiones,
con aumento de la capacidad degranulación del tejido, desplazamiento epitelial para propiciar la
continuidad del tejido y así disminuir el tiempo de solución de la úlcera. Se
pueden realizar angiografías para la revisión de las venas o arterias que se
encuentren obstruidas y así tratarlas de forma adecuada.(3)
Sin embargo la mayoría de las úlceras venosas se
tratan con vendas de compresión, elevación del miembro inferior (ya que
normalmente se suelen encontrar en los miembros inferiores), utilizando
apósitos especiales para favorecer el microambiente que lleve a una mejor
cicatrización yla absorción del líquido
serosanguinolento de la úlcera.
Todos estos mecanismos y la base inmunológica de las
úlceras crónicas se desarrollaran a continuación, debido a que todas de una
forma directa o indirecta intervienen en la respuesta inmunitaria, inhibiéndola,
como en la infección por Mycobacterium ulcerans, causando un aumento de
leucocitos y disminución de la extravasación de estos al sitio de lesión en
donde se encuentre la úlcera afectando de esta manera los procesos de
inflamación de la misma, o incrementándola, como en el caso de las afecciones
vasculares y en desórdenes metabólicos, donde aumenta la acumulación de células
proinflamatorias en el sitio de la lesión.(1) Igualmente se
evidencia en esta patogénesis multifactorial la presencia de fenomenos de
formación de biopelículas (biofilms), que contribuyen a perpetuar la presencia
infecciosa, con automodulación gracias a los fenómenos de Quorum sensing.(6)
Tipos de úlceras
Las úlceras son una discontinuidad de la
barrera epitelial, consiste en una herida convexa de bordes irregulares. Puede
ser superficial afectando la epidermis o profunda dañando tejido nervioso y
muscular, hasta exponer el hueso en el área de la lesión.(7)
Úlceras Vasculares
Heridas causadas por insuficiencias vasculares, donde el flujo sanguíneo
se ve comprometido, concluyendo en isquemia del tejido.
1.- Arteriales
Las
úlceras arteriales también conocidas como úlceras isquémicas, son causadas por
insuficiencias arteriales. Generalmente ocurren en pacientes mayores de 50
años, ocasionalmente son encontradas en pacientes más jóvenes con Diabetes
Mellitus o hiperlipidemias. Son más comunes en hombres, sin embargo, fumar y
algunos factores nutricionales aumentan la incidencia en mujeres.(8)
La
interferencia del flujo sanguíneo puede ser extramural, debido al choque con el
tejido cicatricial (ejemplo: vasculitis o ateroesclerosis), o intramural
(ejemplo: trombosis). El flujo sanguíneo cutáneo se hace inadecuado y no
satisface las demandas metabólicas del tejido local, dañando la piel.
2.- Venosas
En la insuficiencia venosa crónica (IVC), la pared
venosa sufre cambios estructurales y funcionales. El vaso se engrosa y deforma
con aumento en su permeabilidad y pérdida de sus propiedades antitrombóticas.
Como consecuencia de la hipertensión venosa mantenida se altera la microcirculación,
provocando la aparición de lesiones. Además es la causa más frecuente de
úlceras en las piernas, aproximadamente en el 1% de la población.(9)
Los procesos subyacentes a
la IVC, como las zonas cutáneas denominadas dermatosclerosis, culminan en la
degeneración de las células de la piel, del tejido celular subcutáneo e incluso
de la fascia, musculatura, tendones y tejido óseo, y se desarrolla una herida
crónica con escasa tendencia a la curación espontánea.(10)
Las
venas varicosas que aparecen como consecuencia de la insuficiencia del sistema
venoso superficial, presentan cambios estructurales importantes que se asocian
a modificaciones funcionales. En estas modificaciones adaptativas
estructura-función juegan un papel muy importante factores de origen
endotelial, así como interacciones del endotelio con células sanguíneas y con
las células musculares lisas (SMC, del idioma inglés) de la pared del vaso.(9)
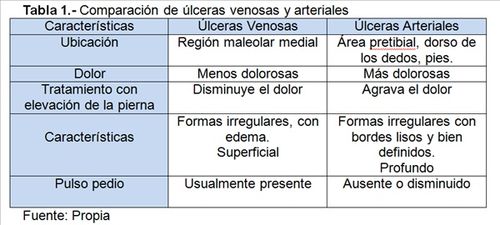
En
la tabla 1 se indican características diferenciales entre úlceras arteriales y
venosas.
En la actualidad se sabe que muchos de los
síndromes asociados a la enfermedad venosa de los miembros inferiores,
trombosis, ulceraciones de origen venoso, entre otros; son en gran medida
responsabilidad de las interacciones entre células sanguíneas y el endotelio
venoso. El estudio ultraestructural de la pared venosa insuficiente muestra
cómo sus células componentes tratan de compensar las necesidades de la pared
ante la hipertensión-hipoxia mantenida.(9)
El endotelio participa en la síntesis de elementos
necesarios para contrarrestar la débil contractilidad del vaso, ayudándolo a
cumplir con el retorno venoso comprometido, sintetizando sustancias tales como
factores vasoactivos, mediadores inflamatorios y componentes del tejido
conectivo.
Desde las primeras etapas de la IVC se producen
cambios funcionales en las células musculares lisas, las cuales proliferan
localmente favoreciendo el engrosamiento de la pared. Estas SMC participan en
la producción de grandes cantidades de tejido conectivo y de enzimas
lisosomales degenerativas.
En
la vena varicosa la regulación del tono venoso dependiente de las SMC está
alterada, pues el desarrollo de tejido conectivo produce grandes separaciones
entre las células rompiendo la distribución armoniosa del influjo nervioso.
Como consecuencia las SMC de la vena insuficiente no pueden producir la debida
contracción de la pared.
Las
células endoteliales gracias a su posición de barrera limítrofe entre la sangre
y el vaso están capacitadas para recibir y responder ante señales de su medio
ambiente que constituyen actividades claves para la vida. Se interconectan con
células de la sangre y el vaso a través de comunicaciones autocrinas y
paracrinas que les permiten controlar el metabolismo de la pared respondiendo
rápidamente a necesidades locales.
Los
leucocitos, especialmente los granulocitos, por ser células altamente reactivas
mantienen intensas interacciones con el endotelio, incluso en condiciones
fisiológicas. La elevada reactividad del leucocito determina su participación
en la fisiopatología de las enfermedades vasculares.(9)
Aproximadamente
la mitad de los granulocitos circulantes en el torrente circulatorio se
encuentra vinculado a la pared de los vasos, principalmente a las venas,las
fuerzas de unión son tan débiles que ruedan siguiendo la dirección del flujo a
lo largo de la pared del vaso. Estas interacciones se realizan mediante
moléculas de adhesión de la familia de las selectinas en los leucocitos y
estructuras de carbohidratos de moléculas de adhesión de la familia de las
mucinas en el endotelio.
Se
ha demostrado que las células endoteliales (CE) activadas por hipoxia
sintetizan y liberan factor activante plaquetario (PAF), un mediador
inflamatorio muy fuerte, cuya acción promueve la adhesión firme del leucocito,
su activación y migración posterior al espacio subendotelial. Las moléculas de
adhesión que intervienen en esta unión firme y en la transmigración del
leucocito son integrinas leucocitarias. En neutrófilos y monocitos esta
interacción está mediada por integrinas de la familia de la B2:
LFA-1; mac-1 y la p150. Adicionalmente en los monocitos también interviene la
integrina VLA-4 de la familia B1 y otras integrinas de la B3.Como
contrarreceptor se encuentran en el endotelio ya activado moléculas de adhesión
de la superfamilia de las inmunoglobulinas tales como ICAM-1 y VCAM-1.(9)
La IVC se caracteriza por variaciones en la
expresión de la integrina CD11b/CD18 (MAC-1) y la L-Selectina. Los niveles de
CD11b disminuyen durante la presencia de la lesión. Los niveles de la
L-Selectina aumentan, indicando la activación de los leucocitos en respuesta a
la hipertensión y su consecuente adhesión al endotelio.(11)
La permeabilidad incrementada de los capilares
dañados promueve la extravasación de proteínas plasmáticas, que sobrepasan la
capacidad reabsortiva de los vasos linfáticos, incrementando el riesgo de
infección. Se producen edemas que limitan la microcirculación de áreas vecinas
por compresión externa de la luz de otros capilares no afectados o por la
acción indirecta de mediadores inflamatorios liberados, disminuyendo el riego
sanguíneo de la zona y así el efecto de barrido de los microorganismos
patógenos que pueden colonizar la lesión característica de la IVC.
Después
de un daño importante en la función capilar en la que interviene tanto el
atrapamiento de leucocitos con la consecuente secreción de productos tóxicos
(citocinas, radicales libres de oxígeno, entre otros.),la formación de fibrina y la formación de
edemas por el incremento en la permeabilidad, se produce isquemia local del
tejido.
Las
alteraciones descritas comprometen tanto a los capilares que deben nutrir al
tejido como a las vénulas poscapilares que deben transportar los productos de
desecho de su metabolismo. Cuando un gran número de vasos capilares está
alterado el defecto nutritivo provoca lesiones tisulares características de la
IVC, la dermolipoesclerosis y la úlcera.(9)
Durante el curso de la enfermedad se
presenta enlentecimiento en la cicatrización de las úlceras, esto se debe
principalmente a la hipertensión venosa, ya que los fibroblastos expuestos al
aumento de la presión venosa presentan una morfología atrófica y la producción
de proteínas específicas que sugieren el envejecimiento prematuro de las
células, además de retraso en su crecimiento y expresión de factores de
senescencia, causando la cronicidad de la lesión y la ausencia de la solución
espontánea de la herida.(12)
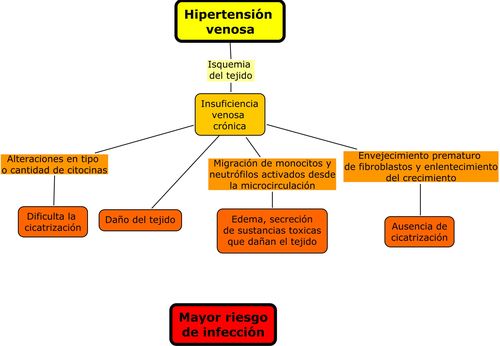
En la figura 1 se esquematiza la
evolución de úlceras vasculares venosas y las relaciones que posibilitan y
favorecen la infección.
Desordenes metabólicos
Pie diabético
Bajo
la denominación de pie diabético han quedado englobados distintos tipos de
fenómenos que, aunque obedecen a distintos mecanismos etiopatogénicos, tienen
de común la localización en el pie de estos pacientes. Existe un acuerdo
universal en los factores determinantes principales: isquemia, neuropatía e
infección. Más de uno de estos factores, y en ocasiones los tres, participan en
su aparición. Se ha estimado que
del 85 al 90 %de todas las amputaciones de causa no traumática se
producen debido a la diabetes y algunas de ellas son debidas a la neuropatía y
a la infección con énfasis en la isquemia (producida por la oclusión de las
arterias más pequeñas por debajo del nivel poplíteo y particularmente las del
pie mismo). La neuropatía puede causar infección y destrucción del antepié,
originando el 20%de las gangrenas diabéticas, mientras que el 80
%restante son debidas a la insuficiencia vascular.
Un
estudio epidemiológico realizado demostró que las tasas de mortalidad yamputaciones
mayores estaban altamente correlacionadas con el tipo de lesión inicial del
pie. Encontrándose particularmente elevadas cuando dicha lesiónconsistía en una gangrena isquémica. A la vezse cuestiona si la lesión nerviosa es metabólica o isquémica, o existe una
suma de factores. Por lo tanto en las personas diabéticas las lesiones
gangrenosas de los dedos son una consecuencia de la isquemia y el mal
perforante plantar depende de otra causa.
La
microangiopatía puede ser responsable de la neuropatía, y su típica expresión
el mal perforante plantar por lesión de los vasa nervorum, y en la
macroagniopatia, por la lesión de los vasa vasorum. El engrosamiento de la
membrana basal de capilar siendo una barrera para el pasaje de las células
blancas del compartimiento vascular a las zonas afectadas favorece la
infección.
En
función de todas las consideraciones anteriores se clasifica el pie diabético,
desde el punto de vista clínico, en dos grandes grupos: el pie diabético isquémico, cuando la lesión inicial es de tipo
úlcera o gangrena isquémica, con independencia de que exista o no infección
sobreañadida; y el pie diabético neuroinfeccioso,
cuando la lesión inicial es de tipo mal perforante plantar, absceso, flemón
difuso o de una asociación de estos, como habitualmente sucede.(13)
Trastornos
inmunológicos:
En los pacientes diabéticos existen alteraciones
inmunológicas las cuales afectan el transporte transcapilar de las células a el
espacio extravascular,acumulándose
granulocitos en la superficie; macrófagos, CD3, CD1, células de Langherhans en
la epidermis y fibroblastos en los márgenes de las úlceras.
También se encuentra el incremento de la expresión de
moléculas de adhesión responsables de la extravasación, las cuales no se
relacionan con el aumento de células inflamatorias en la úlcera por el pie
diabético.
La curación de la úlcera en el pie diabético está obstaculizado
por mecanismos que disminuyen la acumulación de leucocitos, no hay incremento
de la migración de estosy puede haber
un mal funcionamiento de las células de la piel. Las células destinadas para la
cicatrización están disminuidas o anormales y se ven evidenciadas las fases de
la inflamación, que son:
ØRodamiento: empieza cuando los leucocitos se adhieren
de forma laxa a la pared del endotelio por una interacción de baja afinidad de
una selectina y carbohidratos. Las citocinas inducen en el endotelio la
expresión de moléculas de adhesión de las familias de las selectinas E y P que
se fijan a la molécula de adhesión celular similar a mucina en la membrana del
leucocito.
Esta interacción con el endotelio dura poco tiempo por
la fuerza que tiene la sangre circulante que acaba de desprender la célula; por
lo que las moléculas de selectina se ubican una sobre otra célula endotelial, y
además se vuelve a fijar el leucocito fenómeno conocido como rodamiento. Las
principales células que llegan al endotelio y extravasan los tejidos son los
neutrófilos, posteriormente migran los monocitos y por último los linfocitos B
y T.
ØActivación de quimioatrayentes: al final del
rodamiento se activan las citocinas expresadas en la superficie del endotelio y
el repertorio de receptores en leucocitos, proporcionan un grado de
especificidad en el reclutamiento de glóbulos blancos en el sitio de infección.
ØParo y adhesión: las citocinas unidas a su receptor de
superficie en leucocitos da por resultado un cambio de conformación y el
agrupamiento de integrinas sobre el leucocito, permitiendo que se unan con más
firmeza al endotelio, reduciendo la probabilidad de ser arrastrado por la
sangre.
ØMigración transendotelial: los leucocitos se comprimen
para pasar por dos células del endotelio sin alterar la barrera endotelial y
esto se debe a una interacción homotípica entre PECAM1 endotelial y PECAM1
leucocítica.
Por último obtenemos que en el pie diabético hay una
interacción entre las células de la piel y células del infiltrado y que las
primeras células que se encuentran capaces de cicatrizar son los granulocitos,
produciendo aumento de las células de Largherhans.(13)
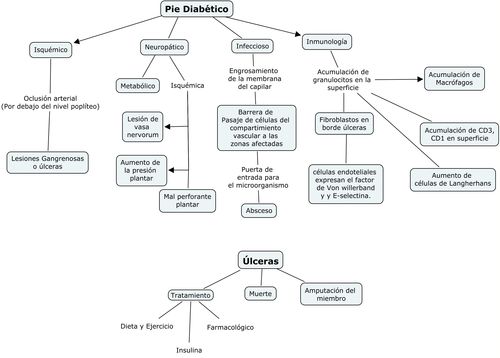
La figura 2, resume la causalidad multifactorial de las úlceras en el
pie diabético.
Figura 2: Etiopatogenia del pié diabético.
Citocinas en úlceras venosas
Los niveles de citocinas inflamatorias
en úlceras por insuficiencia venosa crónica, antes y después de una terapia de
compresión, van a ser clave para la curación de las mismas. Se han realizado
estudios que indican que, elevados niveles de citocinas inflamatorias han estado
implicados en la patogénesis de la no curación y en la insuficiencia de úlceras
venosas crónicas.
Los pacientes sometidos a este tratamiento se designan como rápidos o
con retraso de curación, en base a la superficie de úlcera sanada para un
tiempo determinado.
En la curación rápida, el paciente presenta niveles altos de: IL-1 α y IL-1b(inducción a fiebre, reacción de fase aguda y
estimulación de la producción de neutrófilos), IFN-g (activa la
proliferación celular de células Th1 y regula la expresión de moléculas de MHC
clase I en células nucleadas), IL-12p40 (factor importante en la inducción de
diferenciación del subgrupo Th1 de células cooperadoras, induciendo además la
producción de INF por las células T y células NK, e intensificando la actividad
de las últimas), y granulocitos que secretan factor estimulante de colonias de
macrófagos (GM-CSF) encargado de la diferenciación de linajes de las células granulocíticas
y monocíticas, antes de la terapia de compresión. La IL-1 Ra se ve aumentada y
los niveles de INF-adisminuyeron
significativamente después de la terapia, en los pacientes de rápida curación.(14)
Varias investigaciones sugieren que la
alteración en la curación de las úlceras de la pierna se debe al aumento de los
niveles de mediadores inflamatorios, y no debido a la deficiencia de los
factores de crecimiento, indicando también que la alta concentración de INF-g (citocina proinflamatoria) podría ser la responsable del desarrollo de
la ulceración crónica.(15)
Se ha demostrado que la producción de INF-g induce la
expresión de ICAM-1 en las células endoteliales, monocitos, y las células del
músculo liso. Por lo tanto, la sobreexpresión de las
moléculas de adhesión en la microvasculatura cutánea conduce a una mayor
activación y migración de los leucocitos, lo cual podría estar relacionado con
la actividad de esta citocina. Numerosos estudios demostraron que actúan en
sinergia el INF-g con TNF-α; quizás combinando anti-INF-g y terapia anti-TNF-α sería aún más eficaz para promover la sanación de la úlcera de manera
directa, ésta terapia actualmente se encuentra en período de prueba.
En los pacientes que presentan un retraso en
la cicatrización de la herida (su área de sanación no paso del 40% de
superficie), el perfil de citocinas al principio del tratamiento fue el mismo
que el grupo de rápida sanación pero en concentraciones inferiores, lo cual
lleva a pensar que las altas concentraciones al inicio del tratamiento podrían
ser clave en la recuperación de la misma.
La curación de las úlceras crónicas con
insuficiencia venosa, se asocia con un ambiente de citocinas pro-inflamatorias
antes del tratamiento, que refleja metabólicamente el tejido activo alrededor
de la herida que tiene el potencial de sanar. Los resultados de curación en el
tratamiento con terapia de compresión, apuntan a la reducción de citocinas
pro-inflamatorias y niveles más altos de la citocinas anti-inflamatorios como
la IL-1 Ra.
La mayoría de los pacientes con úlceras en la
pierna y asociados a insuficiencia venosa, experimentarán cicatrizaciónprogresiva durante el tratamiento con
compresión sostenida en las extremidadesde 30 mm Hg o mas, pero la curación suele requerir de 3 a 6 meses de
tratamiento o más. A pesar de los buenos resultados de la terapia en la
curación de la mayoría de las úlceras venosas de la pierna, todavía el
mecanismo responsable de este efecto, no está bien definido.(15)
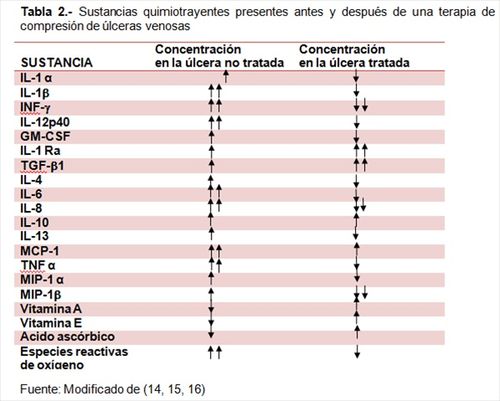
La tabla 2 resume el perfil de citocinas
medidas en las úlceras antes y despues de la terapia compresiva.
El
TGF-b es una citocina mediadora de
la inflamación, secretada por los leucocitos y actúa sobre los fibroblastos que
interviene en el enlentecimiento de la cicatrización de las úlceras, en la
senescencia celular y en el envejecimiento celular prematuro, pero esto no ha
sido completamente comprobado. Esta citocina es un mediador de la fibrosis del
tejido, secretado por los leucocitos y actúa sobre los fibroblastos.(12)
La
citocina antiinflamatoria (IL-10) media la supresión de la respuesta
inmunitaria innata, así como la estimulación de la proliferación de los
queratinocitos mediada por la citocina proinflamatoria (GM-CSF), contribuyendo
con la naturaleza crónica de las úlceras.
Las
citocinas proinflamatorias y antiinflamatorias actúan estimulando o suprimiendo
la respuesta inmunitaria mediada por células, respectivamente, principalmente
actuando sobre las células presentadoras de antígenos. GM-CSF aparece en mayor
grado en las lesiones agudas que en las úlceras y causa una mayor expresión de
moléculas MHC-II en macrófagos y moléculas B7 en células dendríticas,
favoreciendo la presentación antigénica. IL-10 se presenta con mayor frecuencia
en las úlceras, específicamente en el tejido hipertrófico alrededor del sitio
de la lesión, inhibe expresión de moléculas MHC-II en monocitos, inhibiendo la
presentación antigénica y la respuesta inmunitaria resolutiva, causando una
respuesta inmunitaria que no soluciona la lesión si no que favorece su
permanencia.
Las
úlceras causadas por insuficiencia venosa crónica presentan un incremento en la
expresión de IL-10, que suprime la respuesta inmunitaria mediada por células
esperada durante la cicatrización del tejido en condiciones normales.
CD68
es un marcador de superficie presente en células presentadoras de antígeno. Se
presentan en mayor grado en las úlceras por IVC, demostrando así una mayor
presentación de antígenos y una mayor activación de linfocitos, con un
consecuente aumento en la secreción de citocinas. La cantidad de CD68 guarda
relación con la duración de la úlcera.(17)
Úlceras infecciosas
Las
úlceras infecciosas, son aquellas que son causadas por la
colonización de un microorganismo en una herida causando
debilitamiento de la piel y otros tejidos (subcutáneos) y llegando a
causar fibrosis en la lesión.(1)
Úlceras
por micobacterias
Existen
numerosos agentes infecciosos productores de lesiones ulcerosas,
entre los cuales se incluyen
grupos A y G de Estreptococos,
y Pseudomonas
sp., como responsables de piodermas y sepsis. El Staphylococcus
aureus
y la Pseudomonas aeruginosa
pueden estar asociados al
retraso de cicatrización del injerto.(8)
Para
nuestra revisión y posterior elaboración de un modelo de
integración de la génesis de las úlceras crónicas hemos decidido
enfocarnos en una micobacteria,
la cual presentaremos a continuación. A este grupo
se les denomina en ocasiones micobacterias oportunistas o
microorganismos patógenos ocasionales. La mayoría de las especies
pertenecientes a este grupo son casi ubicuas en la naturaleza, y
pueden transformarse en patógenas en circunstancias especiales.
El
Mycobacterium ulcerans, es
un bacilo ácido resistente (BAR) o Ziehl-Neelsen positivo, que crece
muy lentamente en los cultivos entre 6 a 12 semanas y puede tardar
hasta 9 meses en formar colonias visibles en medio sólido.(18) Este
microorganismo es conocido por causar una enfermedad conocida como
úlcera de Buruli; a continuación se presentará una reseña
histórica de la evolución de esta patología.
Se
sabe que después de la aparición de la tuberculosis y la lepra
causadas por dos micobacterias diferentes, apareció una tercera
infección micobacteriana por orden de frecuencia entre los
individuos inmunocompetentes producida por la Mycobacterium
ulcerans. Fue MacCallum quien
describió por vez primera el microorganismo causante al descubrir
bacilos acidorresistentes en una biopsia tomada de una úlcera de la
pierna de un niño en Bairnsdale (Australia) en 1940 y quien publicó
la primera descripción clínica de esta nueva infección
micobacteriana en1948. Sin embargo, la enfermedad ya se conocía en
África antes de 1948 debido a que Sir Robert Cook había descrito en
1897 unas úlceras extensas, causadas casi con toda seguridad por el
Mycobacterium ulcerans.
Entre 1923 y 1935, Kleinschmidt, médico misionero en el nordeste del
Congo, observó también lesiones cutáneas con bordes excavados y
ricas en bacilos acidorresistentes.(19)
Posteriormente
a este descubrimiento se fue investigando la etiopatogenia de esta
enfermedad y se observó que la
infección por M. ulcerans se
transmite por medio del ambiente, a través de pequeñas soluciones
de continuidad (heridas) de la piel. Actuando como puerta de entrada
a través de la cual dicha bacteria puede invadir la dermis y el
tejido celular subcutáneo. Un ejemplo: es la transmisión durante
los trabajos de campo, por medio de su inoculación traumática por
algunos tipos de hierbas o arbustos que puedan contener la
micobacteria. Sin
embargo, la transmisión en el hombre también puede venir dada por
acontecimientos naturales, como las inundaciones, ya que la
Mycobacterium ulcerans
puede crecer en aguas estancadas, así como también debido a
modificaciones del suelo como consecuencia de la acción del hombre
entre ellas las construcciones. Otra
posible forma de infección podría ser a partir de las picaduras de
insectos acuáticos. Los cuales habitan en las raíces de plantas
acuáticas de regiones con climas templados y tropicales, ricos en
agua fresca. Por otra parte, se considera que la transmisión persona
a persona es un fenómeno excepcional. Su
patogenia comienza con su entrada en el tejido cutáneo, donde va
generalmente a producir un nódulo o una pápula cutánea indolora
que en ausencia de tratamiento, evoluciona hacia una ulceración
masiva de la piel y en ocasiones afecta a las extremidades. Esto
sucede como consecuencia de la producción de una toxina necrotizante
(micolactona o mycolactona) por parte del M.
ulcerans, actuando como
inmunosupresora local que tiene afinidad en los adipocitos con efecto
citotóxicos sobre ellos. La necrosis resultante proporciona un medio
que, al favorecer la proliferación del microorganismo causante,
determina la aceleración de la propia necrosis. Durante
esta fase necrótica, la respuesta celular del huésped es muy débil
o inexistente en un determinado momento y por un mecanismo
desconocido, la toxina puede resultar neutralizada, o bien las
bacterias dejan de proliferar y de producirla.
La
curación parece iniciarse cuando el huésped desarrolla una
inmunidad celular contra los componentes del Mycobacterium
ulcerans esto se evidencia por
la formación de unos granulomas los cuales destruyen a las
micobacterias y la enfermedad remite dejando cicatrices.
En
caso de que se observe un elevado número de bacterias extracelulares
y de una escasa respuesta inflamatoria en las lesiones causadas por
Mycobacterium ulcerans sugiere
que los individuos que desarrollan la enfermedad fagocitan de forma
ineficaz o no consiguen fagocitar las micobacterias antes de ser
destruidas, procesadas y presentadas a los linfocitos T.
La
inmunidad celular y del equilibrio en los distintos patrones de
secreción de citocinas en las respuestas Th1 y Th2 juegan un papel
fundamental en el individuo que presenta la úlcera, ya que un cambio
del patrón de citocinas de una respuesta Th1 (IFN-γ, IL-2) a una
respuesta Th2 (IL-4, IL-5, IL-12) y un predominio innato o adquirido
de respuesta Th2 frente a una respuesta Th1 defectuosa podría
facilitar el desarrollo de la enfermedad o condicionar sus
características clínicas y evolutivas. También
se relaciona con mutaciones en los genes que codifican la síntesis
de citocinas Th1 (como por ejemplo la IL-12) y/o sus receptores que
son el receptor del interferón gamma [IFN-γ R] o receptor de la
interleucina 12 [IL-12R]. Las
mutaciones homocigotas de dichos genes dan lugar a un déficit
parcial o completo de la vía de citocinas Th1, el cual es un factor
activador importante de los macrófagos, por consiguiente los déficit
completos dan lugar a infecciones precoces y letales con una escasa
respuesta granulomatosa, mientras que los defectos parciales pueden
manifestarse en edades más avanzadas, presentando un mejor
pronóstico y dando lugar a lesiones con la formación de
granulomas.(19) Los
pacientes con úlcera de Buruli desarrollan una respuesta humoral
inmune frente a M. ulcerans,
evidenciada por la producción de anticuerpos específicos (con un
aproximado de un 70%). Sin embargo, presentan una intensa anergia
cutánea sistémica de células T frente a antígenos de dicha
micobacteria. Desconocemos los mecanismos patogénicos implicados en
dicha anergia, aunque se ha postulado tanto la participación de la
Mycolactona
como de alteraciones inmunológicas y modificaciones en los patrones
de producción de citocinas. Induciendo
inmunosupresión local en el paciente, inhibiendo las respuestas
inflamatorias, detención en el ciclo celular G0/G1, las citocinas
proinflamatorias como TNF-
e IFN-
las cuales se ha demostrado que se expresan durante todas las
diversas etapas de la infección y por último tiene la capacidad de
producir apoptosis de las células.(1) Sin
embargo la
Mycolactona
puede causar la úlcera en sitios lejos de los sitios de infección,
induciendo una respuesta leve inflamatoria, daño de las fibras
musculares, producción de una fibrosis, en donde se ve evidenciado
un aumento de los mastocitos los cuales producen una respuesta
proinflamatoria con una síntesis de citocinas y factores de
crecimiento junto al reclutamiento de los leucocitos en el músculo
esquelético. Cuando
la respuesta inflamatoria ocurre en los sitios lejos de la infección
se crea la proliferación celular y la migración del músculo
esquelético y desgranulación de mastocitos, estos hechos pueden
ocurrir en cualquier momento de las fases en que se presenta la
fibrosis. Normalmente los subconjuntos de los leucocitos neutralizan
la fagocitosis bacteriana y actúan como presentadoras de antígeno a
las células T citotóxicas y helper, pero se a ha demostrado que la
presencia crónica de neutrófilos y macrófagos en el sitio de
lesión explica que no se pudo eliminar el agente causante por lo que
se ve evidenciado la capacidad de
Mycobacterium ulcerans
para inhibir los pasos de resolución de la inflamación.
Una
úlcera se va a desarrollar en tres etapas, inicialmente se empieza
con la presencia de un nódulo, pápula o edéma en el sitio de
lesión correspondiendo a la primera fase de la infección, el cual
es indoloro y pruriginoso. En
ausencia de un tratamiento la lesión va a evolucionando creando una
úlcera en la piel sin bordes definidos la cual corresponde a la
segunda etapa de la lesión. Posteriormente
se empieza la tercera fase que corresponde a una curación espontánea
y formación del granuloma; en algunos casos se puede presentar una
cuarta fase que es la presencia de una fibrosis. En
algunos casos estas lesiones pueden causar complicaciones
incapacitantes como
deformidades por contractura, amputación de miembros y pérdida de
órganos (ojo, mama, genitales). Se han descrito algunos casos de
fallecimiento debido a septicemia, tétanos o hemorragia. También se
ha notificado un número creciente de infecciones óseas que
complican el tratamiento de estos casos. Pueden deberse a la
propagación directa desde la lesión cutánea suprayacente, o
avalar la hipótesis de la diseminación hematógena. (19)Mycobacterium
ulcerans tiene la capacidad de
formar biofilms, definidos como comunidades de microorganismos que
crecen en una matriz de exopolisacáridos y adheridos a una
superficie inerte o un tejido vivo, produciendo a la bacteria una
resistencia contra factores ambientales contrarios y los
antimicrobianos.(6,20)
En
la actualidad no se conoce
exhaustivamente la tasa de morbilidad a nivel mundial y de los
países que presentan la úlcera de Buruli. Por lo general, la
enfermedad afecta sobre todo a los habitantes pobres de zonas rurales
alejadas, en las que el acceso a la atención de salud es limitado
especialmente a los niños menores de 15 años. Ningún grupo racial
o socioeconómico está a salvo.(21)
Recapitulando
sobre la patogénesis toxigénica de la úlcera de Buruli, se han
descrito una familia de moléculas solubles e hidrofóbicas, lípidos
macrocíclicos, estables conocidas como las Mycolactonas
que actúan como toxinas con un efecto tanto citotóxico como
inmunodepresor local, esta penetra en las células por difusión
pasiva y se acumula en el citoplasma inhibiendo así la síntesis
protéica; inducen
la apoptosis de los macrófagos e inhiben tanto la proliferación de
linfocitos T como la producción de interleucina 2 por las células T
y de TNF-α por parte de los monocitos; la anergia observada en la
úlcera de Buruli podría ser debida en parte por la acción de esta
Mycolactona.
Se han implicado, asimismo, otras enzimas, una fosfolipasa C y una
hemolisina (que induciría la producción de TNF-α) que darían
lugar a efectos tóxicos en la infección por M.
ulcerans. No ha podido
establecerse ninguna correlación entre la cantidad o tipo de
micolactona y la intensidad o evolución y forma clínica.
Estas
observaciones parecen sugerir que el número y el tipo de
mycolactonas implicadas podrían influir en el desarrollo de patrones
clínicos distintos. Sin embargo, la participación de otros factores
acompañantes (biológicos, económicos, culturales, disponibilidad
de recursos sanitarios) puede, asimismo, ser fundamental. A
pesar de conocer la acción de la micolactona en el organismo se
desconocen los mecanismos intrínsecos de la respuesta inmunológica
en la infección por M. ulcerans.
La existencia de una anergia de células T frente a M.
ulcerans parece representar un
fenómeno importanteen
el desarrollo de la úlcera de Buruli. Se ha demostradoque las células mononucleares
de la sangre periféricade
los pacientes con enfermedad activa o tras la extirpaciónquirúrgica presentan tanto una
proliferación linfoidecomo
una producción de IFN-γ reducida en respuestafrente a las micobacterias. Esta
anergiaparece
ser adquirida y puede persistir durante años despuésdel tratamiento y resolución de
la lesión. El
IFN-γ es un factor importante en la activación macrofágica y
desempeña un papel fundamental en la protección frente a las
micobacterias. Una baja producción de IFN-γ en pacientes con
infección por M. ulcerans podría
ser, en parte, la causa de la persistencia de las micobacterias
extracelulares, la evolución indolente de la enfermedad y la falta
de respuesta al tratamiento antibacteriológico convencional. Se
ha sugerido que una elevada producción de IL-10 por las células T
con un patrón Th2 podría estar implicada en la baja respuesta de
IFN-γ específica frente a M.
ulcerans. Se han demostrado
perfiles distintos de expresión de citocinas en las distintas fases
de la enfermedad: la presencia de nódulos se asociaría con una
producción mayor de IFN-γ y menor de IL-10, mientras que en las
fases ulceradas se objetivaría unas concentraciones menores de IFN-γ
y una producción superior de IL-10. Una
producción inadecuada de citocinas puede desequilibrar el sistema
inmunitario de los pacientes que desarrollan una infección por M.
ulcerans. En el momento del
desarrollo de la enfermedad se ha descrito un cambio del patrón de
citocinas de una respuesta Th1 a una respuesta Th2, que facilita la
producción de anticuerpos específicos, por lo que han sugerido que
las respuestas Th1 pueden prevenir el desarrollo de la enfermedad.
Un
mejor conocimiento de los mecanismos patogénicos implicados en el
desarrollo de estas lesiones puede permitir en un futuro plantear
posibles intervenciones terapéuticas mediante citocinas que
estimulen una respuesta Th1. Un patrón de producción de citocinas
predominantemente Th2 da lugar a una inhibición de la actividad
macrofágica, y lleva a una disminución de la resistencia contra
diferentes microorganismos. Una respuesta inmunológica de tipo Th2,
que aumentaría la susceptibilidad de los individuos al desarrollo de
úlcera de Buruli, tras entrar en contacto con el agente causal. El
TNF-α podría ser otro factor adicional importante en la activación
de los macrófagos, que fagocitan y destruyen las micobacterias. El
TNF-α es un factor fundamental en la protección frente a
infecciones por micobacterias, esto se evidencio ya que los ratones
deficitarios en TNF-α son altamente susceptibles a la infección, y
no consiguen desarrollar una respuesta granulomatosa.(21) Sin
embargo, no se ha podido demostrar de forma definitiva su papel en el
desarrollo de las lesiones cutáneas, en la trombosis subyacente y en
la necrosis de los adipocitos.(18)
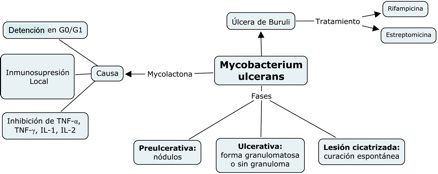
En
la figura 3 se presentan resumidos las fases de la infección por M.
ulcerans y la acción de su
toxina.
Figura
3.- Fases patogénicas de la
infección por M. ulcerans
Elporcentaje de macrófagos y de linfocitos
CD4/CD8 encontrados en el sitio de lesión, junto con su expresión de de IFN-γ,
IL-10, TNF-α Y TGF-β, fue relacionadacon la respuesta inflamatoriaevaluada por histopatología. Todos los casos de úlcera de Buruli
mostraron una necrosis extensiva e inflamación crónica. La característica más
importante fue la presencia o ausencia de granulomas coexistentes junto con un
balance mixto de citocinas pro-inflamatorias/anti-inflamatorias.
Cuando los granulomas estaban
presentes se veía una significativamente alta expresión de IFN-γ; mientras que
en lesiones ulcerativas sin granulomas había un aumento en la expresión de
IL-10 y un mayor contaje bacilar.(21)
Estas características se correlacionan con la
cronicidad de la lesión, las lesiones de mayor duración mostraron granulomas,
Así que, los granulomas estaban ausentes en las lesiones ulcerativas
relativamente tempranas, las cuales conteníanmuchos bacilos y poco IFN-γ, esto sugiere que durante esta etapa de la
enfermedad una fuerte supresión de la respuesta inmune celular facilita
proliferación bacilar.(19)
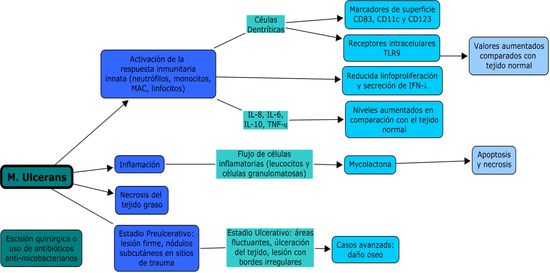
La figura 4 esquematiza la
respuesta inmune en la formación de la úlcera de Buruli.
Figura
4.- Efectos de la infección
por M. ulcerans en el huésped
Por
todo esto se considera que la
reacción inmunitaria del huésped desempeña un papel importante actuando como
factor determinante de la extensión de la lesión y es interesante señalar el
hecho de que en el caso de muchas lesiones, se piensa que estas curan
espontáneamente. La ulterior acumulación (y/o
inducción) de la toxina puede ser lo bastante importante como para provocar la
lisis del macrófago huésped y paralizar las funciones celulares de los
linfocitos y los macrófagos infiltrantes. A su vez, esta inmunosupresión local
podría contribuir a retrasar la reacción inmunitaria sistémica precoz al
antígeno bacteriano, lo que explicaría el hecho de que, a menudo, los pacientes
con lesiones evolutivas no reaccionen a la administración intradérmica de un
antígeno derivado del M. ulcerans (conocida como la prueba de la
burulina).
Más adelante, en la fase de
curación caracterizada por la aparición de granulomas, la conversión de la
intradermorreacción a la burulina a la positividad indica que se desarrolla una
respuesta celular específica. La reacción de la inmunidad celular desempeña un
papel importante en la curación. Cabe destacar queno se observó reacción en las fases iniciales
de la infección por el M. ulcerans, mientras que se obtuvieron
respuestas positivas en los pacientes con lesiones en vías de curación.
Las investigaciones sobre las
reacciones mediadas por anticuerpos frente a las micobacterias no han dado
frutos debido al solapamiento antigénico en este grupo de microorganismos y a
su ubicuidad en la naturaleza. Sin embargo, la patogénesis de la infección por
el M. ulcerans suscita una importante cuestión: si la Mycolactona inhibe el desarrollo de una
reacción inmunitaria eficaz, ¿cómo logra el huésped superar esta situación
para, a la larga, curarse? Una explicación sería que la toxina es neutralizada
por anticuerpos que van produciéndose lentamente. Al poseer una estructura de
macrólido, es improbable que la Mycolactona
induzca la síntesis de anticuerpos.(19)
Úlceras por microorganismos diferentes a
micobacterias.
Algunos
microorganismos son productores de biopelículas o biofilms, figuran entre
ellos Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus,
entre otros.
El ejemplo clásico
y adaptable a la patología ulcerativa crónica es la Pseudomonas aeruginosa un
bacilo gramnegativo no fermentador. Es una bacteria intrahospitalaria oportunista, produce infecciones
cuando el huésped tiene alteradas las defensas, principalmente en pacientes con
quemaduras extensas, leucémicos, transplantados.(22)
Su hábitat son las
fuentes de agua ambientales, piel, sistema respiratorio superior, colon,
respiradores hospitalarios y humidificadores.(23)
Fue aislada en un 18.6% de las veces de úlceras de
pacientes VIH-positivos, procedentes del Servicio de Dermatología del Hospital
Vargas de Caracas.(24)
Entre sus determinantes de
patogenicidad se encuentra la presencia de un pili, y dos toxinas: la
endotoxina, responsable de la fiebre y del shock séptico asociado a la sepsis.23
La exotoxina A, lamás importante, que inhibe la síntesis proteica en las células
hepáticas, corazón, riñón, pulmón y bazo. Inhibe la captación de aminoácidos a
nivel celular.
Dentro de la gran variedad de patologías que produce
pueden mencionarse: endocarditis de válvula nativa y protésicas, neumonía
intrahospitalaria, meningitis, septicemia en neutropénicos y quemados.(23)
Las
úlceras crónicas en las piernas son colonizadas por bacterias saprofiticas,
estas no influyen en la curación de la úlcera; otros microorganismos no se
comportan de la misma manera con respecto a la formación de tejido sano.(25,26)
P.
aeruginosa ocasiona crecimiento de la herida, a pesar del tratamiento
antimicrobiano. Se demostró que
el deterioro de la úlcera fue causada por la invasión al tejido circundante,
causando el crecimiento de la ulcera y la formación de anticuerpos. La
exotoxina A ejerce efectos patogénicos en el retraso de la curación de las
heridas y restauración de los procesos normales de curación con anticuerpos de
exotoxinas A. Sin embargo, el estudio señaló que la cantidad de exotoxina no
está relacionada con el tamaño de la úlcera.(25)
LLLLos biofilms son
grupos de bacterias mantenidas en sustanciapoliméricaextracelular asociados
a una superficie responden parcialmente los antimicrobianos. Sin embargo, al
ubicarse en espacio de la herida causan cronicidad y perpetúan la inflamación
asociada.
Las hipótesis que
explican la permanencia de los biofilms plantean que el huésped desarrolla una
respuesta hiperinflamatoria incapaz de superar la barrera, nutre las bacterias
y las perpetua, convirtiendo la herida en crónica. El biofilm interactúa conel huésped en un enlace estable de nutrición
en forma de relación parasitaria.(6,27)
El biofilm
secuestra el sistema inmunológico del hospedador siendo capaces de estimular
las células, del hospedador,para
producir citocinas en exceso dañando los tejidos. Por esta razón estas
infecciones corresponden a la ruptura de los conceptos de universalidad(inmensa cantidad de respuestas inmunes pueden generarse en contra de
cualquier reto potencial), tolerancia (barreras mayores que previenen o limitan
la respueta alo propio y a los no
patógenos), apropriateness (respuestas
que tienen solo como blanco a los patógenos. La magnitud de la respuesta
es apropiada con el nivel de amenaza, haciendo un esfuerzo por no causar daño
en el huésped),por los cuales se
rige el sistema inmunológico.
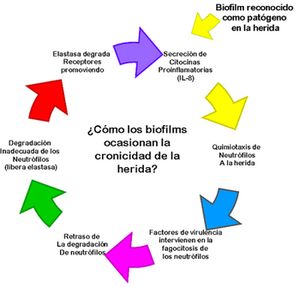
La detección de
biofilm en el organismo estimula la secreción de NF-kB lo que se reconoce por
el huésped como señal de alerta por invasión patógena. El sistema inmunológico
responde ante esta señal con la migración de neutrófilos al área de la herida,
promueven la liberación de citocinas proinflamatorias GM-CSF, IL-1, TNF-a, IFN-g e IL-8.27 Lo anterior se expresa en
forma resumida en la figura número 5.
Figura 5.- Secuencia de eventos donde los biofilms ocasionan la
cronicidad de la herida
La literatura indica un exceso de neutrófilos acumulados en el sitio de
las heridas crónicas y otras infecciones crónicas. La IL-8 en la herida
promueve la quimiotaxis de los neutrófilos, en un sistema inmunológico sano,
estos inician la apoptosis luego de engullir al patógeno, expresando marcadores
de superficie que son reconocidos por los macrófagos. Los productos bacterianos
como los lipopolisacáridos interfieren en la expresión de estas moléculas de
superficie evitando así el inicio de la apoptosis de los neutrófilos y de la
fagocitosispor parte de los macrófagos.
La degradación posterior ocasiona la liberación de proteinasas y
elastasa que interfiere en la cicatrización degradando el tejido de la matriz.
La elastasa degrada los receptores del neutrófilo CXCR1 cuyos fragmentos
estimulan receptores TLR2 a producir más citocinas inflamatorias, ocasionando
la cronicidad de la herida.
Las
heridas crónicas infectadas deben ser desbridadas quirúrgica o enzimáticamente
para eliminar el tejido necrosado y la película bacteriana. Los antisépticos
tienen una limitada eficacia y la resistencia a antibióticos es 1000 veces
mayor para las bacterias de la película que en bacterias de fase planctónicas.
Las bacterias se comunican mediante un sistema de señales químicas (señales quorum sensing) para iniciar la síntesis
de biofilm y de factores de virulencia (6). La interferencia de
estas señales ésta siendo considerada en los tratamientos de las infecciones
crónicas. En previos estudios básicos se ha determinado que Lactobacillus
plantarum y sus productos inhiben tanto in
vivo como in vitro la capacidad
patogénica de P. aeruginosa. En
estudios clínicos preliminares demostramos que L. plantarum es efectivo
en el tratamiento de úlceras y quemaduras infectadas.(28)
Otras úlceras
1.- Por presión
Es un área
de la piel que sufre daños cuando una persona permanece en una sola posición
por mucho tiempo sin desplazar su peso, de un punto fijo de apoyo. Esto con
frecuencia sucede cuando la persona está postrada a una silla de ruedas o a una
cama aún por un corto período de tiempo (por ejemplo, después de una cirugía o
lesión). La presión constante de fuerzas de protuberancias óseas contra la piel
resultan en isquemia y necrosis de los tejidos, formando una úlcera.(26,29)
Las úlceras crónicas por presión están pobremente vascularizadas, con
tejido fibroso grueso y cicatrizial alrededor del borde y queratinocitos
incapaces de proliferar o migrar en los márgenes de la úlcera. Los fibroblastos son escasos y
parecen morfológicamente inactivos. Las heridas y las úlceras crónicas
mostraron diferencias en la deposición del colágeno en el lecho de la herida.
En las heridas que sanan se notan fibras de colágeno madurando ubicadas debajo
de un epitelio nuevo al contrario las heridas crónicas mostraron solo una
pequeña y relativamente desorganizadas fibras de colágeno en el borde.
El
lecho de la úlcera presenta un número variable de vasos y algunos se
encontraron trombosados. Los neutrófilos tendieron a estar localizados en la
pared del vaso cerca de la superficie de la herida. Los monocitos y los macrófagos
se encontraban regados por toda la extensión de la úlcera.
Dentro
de los tratamientos se incluyen cojines y colchones antiescara que mantienen la
circulación del aire evitando la formación de un punto de presión óseo contra
la piel. Un estudio realizado en pacientes de 70 años de edad durante 28 días
demostró que el uso de Factor BB recombinante derivado del crecimiento
plaquetario (en inglés, PDGF). Este factor es capaz de estimular la
proliferación, quimiotaxis y activación de los genes de macrófagos y
fibroblastos, induce la deposición acelerada de matriz y subsecuente formación
de colágeno en modelos experimentales de reparación de tejidos. También
estimula la neovascularización de los tejidos isquémicos.
2.- Hematológicas
Las hemoglobinopatías son deficiencias de los eritrocitos en los cuales
está involucrada la hemoglobina, proteína encargada de fijar, transportar y
liberar oxígeno desde los pulmones hacia los tejidos.(30)
Este tipo de úlceras son isquémicas y necróticas, causadas por la falta
de irrigación del tejido, la diferencia con las úlceras de presión radica en
que la isquemia no se debe a fuerza mecánica ejercida contra el vaso. Debido a
la falta de irrigación sanguínea de los tejidos es necesario que los pacientes
eviten traumas y mantengan una adecuada limpieza de la herida ya que esta
condición sanguínea fundamenta la expansión rápida de la úlcera.
A forma de tratamiento se recomiendan inyecciones subcutáneas locales
del Factor de estimulación de las colonias Granulocito-Macrófago (en inglés,
GMCSF) de actividad múltiple dentro de las que se encuentran el aumento de
IL-1, TNF-a y la disminución de IL-8, mediador de la inflamación.
También activa neutrófilos mejorando la depuración de las bacterias,
promoviendo la neovascularización, aumento de queratinización y por último
acelerando el proceso de formación de tejido nuevo.(31)
3.- Inmunológicas
Las
úlceras inmunológicas son aquellas que tienen como causa deficiencias o errores
del sistema inmune del hospedador, tanto de regulación, como genéticas.
CD59 es
una glicoproteína capaz de prevenir la formación del complejo de ataque de
membrana formado por los complementos terminales de las proteínas C5b a C9, en
la superficie de la célula. Este consiste en el último paso en la apoptosis de
las células por vía de activación del complemento, llevando a cabo la acción de
protección de la célula de la lisis mediada por complemento.(32)
La
respuesta inflamatoria se ve atribuida a la formación del complejo de ataque de
membrana (en inglés, TCC) y su efecto citolítico. La pérdida de esta
glicoproteína aumenta la deposición del complejo terminando con la inflamación
como un factor importante en el proceso de daño de tejidos en úlceras crónicas
de miembros inferiores.(2) Las úlceras isquémicas presentan menos
CD59 que las úlceras por hipertensión venosa.
La
pérdida de reguladores del complemento de la membrana, acompañada con la
activación del complemento se sugiere involucrada en el proceso fisiopatológico
en el daño del tejido en isquemia del miocardio.
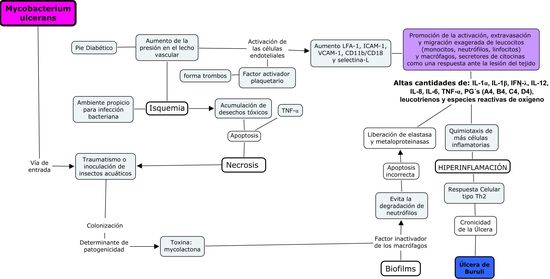
Modelo de integración entre la patología vascular, inmunológica e infecciosa
Luego de una extensa revisión de la literatura existente planteamos una
relación entre los factores vasculares, inmunológicos e infecciososque predisponen a la aparición de una úlcera
crónica. Las úlceras crónicas se inician por medio de una lesión del lecho
vascular que causa la isquemia y necrosis del tejido. La isquemia ocurre por el
aumento de la presión vascular (venosa o arterial). El aumento de la presión
desarrolla un microambiente que induce la activación de las células
endoteliales y el sucesivo aumento de expresión de las moléculas de adhesión
celular: LFA-1, ICAM-1, VCAM-1, CD11b/CD18 y selectina-L, promoviendo la
activación, extravasación y migración exagerada de leucocitos (monocitos,
neutrófilos, linfocitos) y macrófagos como una respuesta ante la lesión del
tejido. Adicionalmente el endotelio secreta factor activador plaquetario
promoviendo la formación de trombos co-causando anoxia. Las células extravasadas, causan la acumulación de citocinas IL-1a, IL-1b, IFN- g, IL-12, IL-8, IL-6, TNF-a,
prostaglandinas de generación tipo 4 (A4, B4, C4, D4), leucotrienos y especies
reactivas de oxígeno. En condiciones normales, estas no serian consideradas
tóxicas para el organismo, sin embargo debido a la reacción exagerada, se
acumulan en grandes cantidades creando un ambiente hiper-inflamatorio. Las
sustancias secretadas nuevamente, inducen la quimiotaxis de más células
inflamatorias no específicas, aumentando aun más la inflamación y mediadores
químicos provenientes de las células involucradas amplificando la reacción de
forma cíclica.
El pie diabético se caracteriza por causar un aumento de la presión en
los vasos de mediano y gran calibre, ocluyendo la luz vascular. La isquemia
crea un ambiente óptimo para la colonización bacteriana anaerobia. La hipoxia
es la fuente de la acumulación de desechos tóxicos que causan la necrosis del
tejido que junto al TNF-a promueven la apoptosis de las células. La
discontinuidad de la barrera epitelial y el microambiente anaerobio promueve la
colonización por las bacterias oportunistas como M. ulcerans y P. aeruginosa. La barrera epitelial es la primera línea de defensa contra los
microorganismos invasores, la necrosis del tejido concluye en la disolución
parcial de esta barrera, sirviendo como puerta de entrada a bacterias
colonizadoras. Debido a la isquemia producida por diversos factores la herida
constituye un nicho óptimo favoreciendo la permanencia, desarrollo y
reproducción del microorganismo invasor.
La mycolactona de M. ulcerans se
especializa en la inactivación de linfocitos y macrófagos, produce una
respuesta de hipersensibilidad tipo IV y síntesis de anticuerpos, causando la
necrosis del tejido yun proceso de
inflamación crónica que va a evitar la cicatrización del tejido.
Los biofilms al igual que la mycolactona evitan la degradación de los
neutrófilos, persistiendo estos por más tiempo que el habitual, promoviendo una
apoptosis errónea. Estos son degradados de manera inadecuada liberando enzimas
líticas metaloproteinasas y elastasa, que al ser liberadas en el área
incorrecta, dañan el tejido circundante. Aumentando la activación de las
células, amplificando adicionalmente el reclutamiento de nuevas células
proinflamatorias, que secretaran más citocinas.
Se evidencia que una elevada producción de IL-10 por Th2, se encuentra
implicada en la baja respuesta de IFN-g específica
frente a M. ulcerans. Esta producción
inadecuada de citocinas desequilibra el sistema inmunitario de los pacientes
infectados con M. ulcerans, en donde
se desarrolla la úlcera de Buruli.
El modelo que integra los diferentes factores patogénicos presentados
anteriormente se presenta a continuación en la figura 6.
Figura 6.- Modelo de integración entre la
patología vascular, inmunológica e infecciosa.
A MANERA DE CONCLUSIÓN
La cronicidad de una lesión se debe a una
respuesta exacerbada que se torna dañina para el tejido o para el huésped en
general. En muchos casos la permanencia de una úlcera es causada por una
respuesta inmunitaria que no logra eliminar, suprimir o controlar el factor causal.
Durante una respuesta ante una úlcera crónica
el perfil de citocinas predominante es: valores aumentados de IL-8, IL-1a y TGF-b, IL-12, INF- g, TNF-a, características de la
respuesta de tipo Th2, es decir una respuesta principalmente humoral, en la que
se activan células plasmáticas y por ende secreción de anticuerpos, que muchas
veces agravan la lesión atacando al tejido del hospedador e impidiendo su
cicatrización.
En el caso contrario, cuando se evidencia una
mejoría, la respuesta es dirigida principalmente por las células Th1, es decir,
una respuesta netamente celular, con la participación de las citocinas IL-10 y
TNF-g, que suprimen la respuesta de las células Th2.
El tratamiento ideal para resolver este tipo
de lesiones seria aquel que busque controlar y lograr una estabilización de la
respuesta de células Th2sin suprimirla
por completo y así atenuar los síntomas de la inflamación crónica, favoreciendo
la cicatrización del tejido y la desaparición de la lesión.
Durante la respuesta inmunitaria ante este
tipo de situaciones se liberan gran cantidad de metabolitos proinflamatorios
causando: aumento de la permeabilidad vascular,acumulación de células del sistema inmunitario, contracción del músculo
liso, disminución del diámetro de la luz de los vasos, aumento de la expresión
de moléculas de adhesión celular, aumento de secreción de sustancias que se
tornan tóxicas para el organismo cuando se presentan en exceso, presencia de
fibroblastos atrofiados y finalmente edema.
En úlceras de diferentes etiologías se
observa como factor común que impide la cicatrización, el factor inactivador de
los macrófagos, que inhibe la producción de ciertas citocinas,por lo que se debe lograr que la
medicaciónpromueva la activación de los
macrófagos y regule la respuesta llevándola a condiciones normales, caracterizada
por la producción de factores que activan a los macrófagos con la consiguiente
degradación del patógeno o de las células que están respondiendo de manera
agresiva contra el huésped.
Referencias
1.Houngbédji MG, Boissinot M, Bergeron GM,
Frenette J. Subcutaneous
injection of Mycobacterium ulcerans causes necrosis, chronic inflammatory
response and fibrosis in skeletal muscle. Microb. Infec. 2008;10(12-13):1236-43. Disponible en: http://www.labmeeting.com/paper/28486073/houngb%C3%A9dji-2008-subcutaneous-injection-of-mycobacterium-ulcerans-causes-necrosis-chronic-inflammatory-response-and-fibrosis-in-skeletal-muscle
2.Abejón-Arroyo A. Simposio: Úlceras de
Etiología Vascular. Tratamiento local
de las úlceras vasculares Angiología 2003; 55 (3): 272-79. Disponible
en: http://www.revangiol.com/pdf/Web/5503/bi030272.pdf
3.Tratamiento de las úlceras vasculares En:
Úlceras vasculares (Editorial) Disponible en: http://www.ulceras.net/monograficos/ulcVasculares08.htm
4.. Kindt TJ, Goldsby
RA, Osborne BA. Inmunología de Kuby. Sexta
edición. México Editorial Mc Gram Hill. 2007
5.Schuette D, Um-Boock A,
Mensah-Quainoo E, Itin P, Schmid P, et al.Development of highly
organized lymphoid structures in buruli ulcer lesions after treatment with
rifampicin and streptomycin. PLoS Negl Trop Dis 2007;31(1):e2 Disponible en:
http://www.ncbi.nlm.nih.gov/sites/entrez
6.Miller MB, Bassler BL. Quorum sensing en
bacterias. Annu Rev Microbiol. 2001;55:165-99 Disponible en:
http://www.ncbi.nlm.nih.gov/pubmed/11544353
7.Harper`s Collins de bolsillo,
diccionario medico,ed. en español Madrid,Marbàn, Dox I, Melloni J, Eisner
G,Melloni J; 2006
8.Micheletti G. Ulcers
of the lower extremities En: Gogia, PPClinical Wound Management. Edition: Illustrated Pub Inc, USA, Publicado
por: SLACK Inc, 1995 p.99-114 Disponible en:
http://books.google.co.ve/books?id=OiP6U3zCb3oC&printsec=frontcover&dq=Clinical+Wound+Management
9.Ramos L, Pérez L, Díaz A, Mahía M. Eventos celulares en el desarrollo de la insuficiencia venosa crónica. Instituto
Nacional de Angiología y Cirugía Vascular.. 2001 Rev Cubana Angiol y Cir Vasc
2001;2(2):142-48. Disponible en http://bvs.sld.cu/revistas/ang/vol2_2_01/ang11201.htm
10.Rodríguez-Piñero, M. Epidemiología, repercusión, sociosanitaria y
etiopatogenia de las úlceras vasculares. Simposio sobre Diagnóstico y
Tratamiento de las Úlceras de Etiología Vascular. 2003.Angiología 2003; 55
(3): 260-267Disponible en: http://www.angiologia.es/pdf/Web/5503/bi030260.pdf
11.Saharay M, Shields D, Porter J, Scurr J,
Coleridge P. Leukocyte activity in the microcirculation of the leg in patients
with chronic venous disease.J Vasc Surg. 1997 ;26(2):265-73. Disponible
en: http://www.ncbi.nlm.nih.gov/pubmed/9279314
12.Stanley A, Fernandez N, Lounsbury K, Corrow
K, Osler T, Healey C, et all. Pressure-Induced Cellular Senescence: A
Mechanism Linking Venous Hypertension to Venous Ulcers.J Surg Res.
2005 Mar;124(1):112-7 Disponible en:
http://www.ncbi.nlm.nih.gov/pubmed/15734488
13.Mc Cook J, Montalvo J, Ariosa MC,
Fernández P. Hacia una llamada clasificación del Pie Diabético 1979. Disponible
en: http://scholar.google.es/scholarías_q=pie+diabetico&num=10&btnG=Buscar+en+Google+Acad%C3%A9mico&as_epq=&as_oq=&as_eq=&as_occt=any&as_sauthors=&as_publication=&as_ylo=&as_yhi=&hl=es&lr=
14.Simka M. A potential role of interferon-c in the pathogenesis of venous leg ulcers. Med. Hyp. 2006; 67: 639644. Disponible en: http://intl.elsevierhealth.com/journals/mehy
15.Beidler SK, Douillet CD,BerndtDF,Keagy BC, RichPB and MarstonWA. Inflammatory
cytokine levels in chronic venous insufficiency ulcer tissue before and after
compression therapy.J.Vasc.Surg 2009; 49:1013-20. Disponible en:http://linkinghub.elsevier.com/retrieve/pii/S0741521408019812
16.WlaschekM, Scharffetter-Kochanek K. Oxidative
stress in chronic venous leg ulcers. Wound
Rep Reg 2005;13:452461. Disponible en:
http://www3.interscience.wiley.com/journal/118664263/abstract?CRETRY=1&SRETRY=0
17.Ya-Qi Li, Doyle J, Roth T, Dunn R, Lawrence
WT. IL-10 and GM-CSF Expression and the
Presence of Antigen-Presenting Cells in Chronic Venous Ulcers.J Surg Res. 1998 Oct;79(2):128-35.Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/9758727
18.Ferrán
M, Coll A,Pujol R. Úlcera de Buruli.
Piel 2005; 20(5):223-30. Disponible en:http://www.elsevier.es/watermark/ctl_servlet?_f=10&pident_articulo=13074567&pident_usuario=0&pcontactid=&pident_revista=21&fichero=21v20n05a13074567pdf001.pdf&ty=78&accion=L&origen=elsevier&web=www.elsevier.es&lan=es
19.Asiedu K, Scherpbier R, Raviglione
M.Organización Mundialde
la Salud. Úlcera de Buruli: Infección por el Mycobacterium ulcerans, 2000, p.6-37. Disponible en:
http://whqlibdoc.who.int/HQ/2000/WHO_CDS_CPE_GBUI_2000.1_spa.pdf
20.Lasa I, Pozo JL, Penadés JR, Leiva J. Biofilms
bacterianos e infección. Anales Sis San Navarra. [periódico en
la Internet]. 2005 Ago [citado 2009 Mayo 26] ; 28(2):
163-175. Disponible en: http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S1137-66272005000300002&lng=es&nrm=iso.
21.Kiszewski A, Becerril E, Aguilar L, Kader I, Myers W, Portael F, Hernàndez
R. The local immune response in
ulcerative lesions of Buruli disease. Clin. Exp. Immunology 2006; 143:44551.Disponible en:
http://www.pubmedcentral.nih.gov/picrender.fcgiíartid=1809619&blobtype=pdf
22.López S,Tuhay N, Gauna A, Ramírez H.Pseudomonas
aeruginosa, Resistente a betalactámicos e inhibidores de betalactamasas
(Piperacilina/Tazobactam).Rev. posgrado VIa. Cátedra Med 2002; 121 Disponible en: http://www.med.unne.edu.ar/revista/revista121/pseudomonas.htm
23.Levinson, W.
Resúmenes de los microorganismos médicamente importantes En: Microbiología e
inmunología médicas. (8va edición), Mexico Ed. McGraw-Hill interamericana, 1999
p 482-522
24.Marcano L MJ,
Urrestazaru MI, Serrano N. Bacterias aerobias aisladas de úlceras cutáneas en
pacientes VIH positivos. Sección de Microbiología, Instituto de Biomedicina
años 2001-2002. Rev Soc Ven Microbiol 2003;23(2): 118-23. Disponible en: http://www.med.unne.edu.ar/revista/revista121/pseudomonas.htm
25.Danielsen L, Westh H, Balsev E, Rosdahl VT,Döring G. Pseudomonas aeruginosa exotoxin A
antiibodies in rapidly deteriorating chronic leg ulcers. Lancet 1996; 347: 265 Disponible en:
http://www.ncbi.nlm.nih.gov/pubmed/8551911
26.Pierce G, Tarpley J, Allman R, Goode P,Serdar C,Morris B, et al. Tissue Repair Processes in
Healing Chronic Pressure Ulcers Treated with Recombinant Platelet- Derived
Growth Factor BB. Am J Pathol 1994:145(6):1399-1409
Disponible en:
http://www.pubmedcentral.nih.gov/articlerender.fcgiíartid=1887508
27.Wolcott R, Rhoads D, Dowd S. Biofilms and
inflammation of chronic wounds. J Wound Care. 2008 Aug;17(8):333-41
Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/18754194
28.Ghanem C, Huaman M,
Molina S,Peral M, Ramos A. Estudios de la importancia del biofilm
bacteriano en úlceras y heridas crónicas. Uso de la bacterioterapia como
tratamiento alternativo Disponible
en:http://www.ct.unt.edu.ar:2000/Helpers/ver_proyecto_ua.aspxéserver=www.ct.unt.edu.ar&id=4940
29.Dox IG, Melloni J,
Eisner GM,Melloni JL. Escara de cúbito En: Harper`s Collins
de bolsillo, Diccionario médico, lugar, editorial, 2006 p. 319.
30.Servicio del Plan de Salud e
Investigación (España) Úlceras de presión En: Guía de práctica
clínica. La prevención de las llagas por contacto, guía para el paciente.
Disponible en: http://www.gobiernodecanarias.org/sanidad/scs/su_salud/ahcpr/guia16-1.gif
31.Voskaridou E,
Kyrtsonis MC, Loutradi-Anagnostou A, Loukopoulos D. Healing of Chronic Leg Ulcers in the Hemoglobinopathies With Perilesional
Injections
34.Baslev E, Thomsen HK, Danielsen L, Sheller J, Garred P. The terminal
complement complex is generated in chronic leg ulcers in the absence of
protectin (CD59). APMIS 1999; 107(11):997-1004. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/10598871
NOTA:Toda la información que se brinda en este artículo es de cará
cter investigativo y con fines académicos y de actualización para estudiantes y profesionales de la salud. En ningún caso es de carácter general ni sustituye el asesoramiento de un médico. Ante cualquier duda que pueda tener sobre su estado de salud, consulte con su médico o especialista.