María Concepción Páez
Doctora en Nutrición. Prof. Asociado Escuela de Bioanálisis. Investigadora y Directora Instituto de Investigaciones en Nutrición (INVESNUT-UC) Universidad de Carabobo.
Anna M. Cioccia
Magister en Nutrición. Prof. Titular Jubilada del Postgrado en Nutrición de la Universidad Simón Bolívar.
Patricio Hevia
phevia@usb.ve
Ph.D. en Nutrición. Prof. Titular Jubilado del Postgrado en Nutrición de la Universidad Simón Bolívar.
Hematología Papel de la hepcidina y la ferroportina en la regulación hormonal de la homeostasis del hierro. (Revisión) Fecha de recepción: 23/07/2014
Fecha de aceptación:
30/08/2014
El hierro es un nutriente esencial y a la vez un elemento potencialmente tóxico. Una particularidad del hierro es que a diferencia de los demás nutrientes la capacidad del organismo para excretarlo es muy limitada y no está sujeta a ninguna regulación, por lo que el exceso del hierro no puede eliminarse. Por esta razón, la homeostasis del hierro depende exclusivamente del hierro absorbido que aumenta o disminuye dependiendo de las necesidades de este metal. Durante el siglo XX se propuso que la mucosa intestinal era capaz de detectar las necesidades de hierro y regular su absorción, sin embargo, los mecanismos celulares que sustentaban esta capacidad de la mucosa se desconocían. En este aspecto, el decubrimiento del sistema hepcidina-ferroportina, en los primeros años del siglo XXI y los avances en el conocimiento de su importancia en la regulación de la homeostasis del hierro, han sido determinantes. La hepcidina es una hormona polipeptídica de 25 aminoácidos que se sintetiza en el hígado y cuyo blanco es la ferroportina, que es un transportador cuya función es extraer al hierro fuera de la célula. Este exportador de hierro, es una proteína transmembrana que se expresa en las células de todos los tejidos que movilizan hierro. En estas células, la exportación de hierro es proporcional a la concentración de ferroportina. La ferroportina es el receptor de la hormona hepcidina y cuando esta se une al receptor, provoca su internalización y degradación intracelular con lo que se reduce su expresión en la membrana y en consecuencia la capacidad de las células de exportar hierro. Un exceso de hepcidina produce un secuestramiento de hierro en las células absortivas de la mucosa intestinal, así como en los macrófagos, reduciendo la absorción de hierro y la capacidad de los macrófagos de liberar el hierro producto de la eritrofagositosis. El resultado es una disminución del hierro sérico y de la generación de glóbulos rojos en el tejido eritropoyético. En contraste, cuando los niveles de la hormona hepcidina disminuyen, la absorción de hierro, así como sus niveles séricos y la capacidad de reciclaje de hierro aumentan. Hay dos condiciones que aumentan la producción hepática de hepcidina. La primera es un aumento sistémico del hierro y la segunda es la inflamación secundaria a la infección o trauma. Esto se debe a que una elevación del hierro sérico y en las células hepáticas, activa la vía de señalización BMP-SMAD, que en el núcleo estimula la transcripción del gen de la hepcidina (HAMP), mientras que la inflamación produce citoquinas inflamatorias que en el hígado activan tanto la vía BMP-SMAD, así como la vía de señalización JAK-STAT que tienen el mismo efecto sobre HAMP. Por lo tanto, en ambos casos los hepatocitos producen más hepcidina, con el consecuente efecto negativo sobre la expresión de la ferroportina, disminuyendo la absorción del hierro dietario, así como la liberación del hierro celular. Esto previene una sobrecarga de hierro o reduce la disponibilidad del hierro a microorganismos invasores. En contraste, la anoxia, en conjunto con la eritropoyetina y factores producidos por el propio tejido eritropoyético, en condiciones en que aumenta la producción de glóbulos rojos, inhiben la transcripción de HAMP y la producción de hepcidina, con lo que la ferroportina exporta libremente el hierro celular. Sin embargo, cuando la estimulación de la transcripción del gen HAMP (alto hierro o productos de la inflación) o su inhibición (anoxia, factores eritropoyeticos) cesa, la producción de hepcidina vuelve a su condición basal permitiendo mantener una apropiada homeostasis del hierro. No obstante, si la producción de hepcidina permanece anormalmente elevada o disminuida se producen alteraciones en el metabolismo del hierro como son anemias refractarias y acumulaciones patológicas de hierro, respectivamente. En esta revisión se describen los aspectos más importantes de la relación de la hormona hepcidina-ferroportina en la homeostasis del hierro, enfatizando los aspectos moleculares que intervienen en su síntesis y producción hepática de la hepcidina. Así como las patologías relacionadas con la sobreproducción o deficiencias en la generación de esta hormona.
Palabras Claves:Hepcidina; ferroportina; regulación hormonal; metabolismo del hierro; exceso de hierro; inflamación; anoxia; anemia crónica; hemocromatosis
Title Hepcidine and ferrroportine in the hormonal regulation of iron homeostasis. (Review)
Abstract
Iron is both an essential nutrient and a potential toxicant and it is unique among the nutrients because its excretion is minimal, not regulated and incapable of removing excessive iron. Therefore, iron homeostasis depends exclusively on absorption. During the 20th century the capacity of the organism to increase or decrease iron absorption according to needs was associated with the ability of the intestinal mucosa of sensing systemic iron and regulating absorption. However the cell mechanism behind this mechanism was unresolved. The system hepcidin-ferroportin discovered at the beginning of the 21th century has contributed in explaining this mechanism. Hepcidine is a 25 aminoacids polypeptide hormone synthesized in liver cells whose target is the ferroportin receptor. Ferroportin is a transmembrane protein, which is the only iron exporting transporter in iron mobilizing cells. In these cells the iron extruded is proportional to the expression of ferroportin on their surface membrane. When hepatocyte produced serum hepcidine, reaches its target, the hepcidine-ferroportin complex is interiorized and degraded in the cell lysosomal system, reducing ferroportin and the cell capacity of exporting iron. As a result, iron absorption as well as iron recycling and serum iron are reduced. In contrast, when the level of the hepcidine hormone decreases, iron absorption, recycling and serum iron increase. Two conditions stimulate hepatic hepcide synthesis and secretion. The first is an elevation of systemic iron and the second is inflammation. High iron is sensed in hepatocytes and it stimulates the BMP6-SMAD cell signaling system, which in the nucleus activates the transcription of the hepcidine gene (HAMP). Inflammated tissues produce inflammatory cytokine production which in liver, stimulate HAMP transcription by activating both BMP6-SMAD and the JAK-STAT signaling systems. This, in both cases result in higher hepcidine which acting on ferroportine, causes lower iron absorption as well as cell sequestration, preventing iron overload or iron availability to invading microorganisms respectively. In contrast, anoxia and stimulated erythropoiesis, down regulate liver hepcidine production by a negative effect of erithropoietin as well as other erythron produced signaling products, on the HAMP gene. When HAMP regulatory factors (high iron, inflammatory products, erythropoietin) dissipate, liver hepcidine production comes back to normal, allowing for proper iron homeostasis. However, if liver hepcidine production remains elevated or abnormally low, alterations in iron metabolism such as refractory anemias or iron overload are produced, respectively. This revision emphasizes the molecular events involved in hepcidin synthesis and the main health problems associated with hyper and hypo hepcidinemia.
Key Word Hepcidine; ferroportine; hormonal regulation; iron metabolism; iron excess; inflammation; anoxia, chronic anemia; hemochromatosis
Papel de la hepcidina y la ferroportina en la regulación hormonal de la homeostasis del hierro. (Revisión)
Introducción
La hepcidina (Hpc) es un péptido que fue
aislado de orina humana por Park y colaboradores en el 2001(1) y
que recibió su nombre con base a su lugar de síntesis (hepatocitos) y a su
comprobada propiedad antibacterial in vitro (-cidina). En el curso de estos estudios, uno de los donantes de
sangre desarrolló una infección sistémica y la concentración de hepcidina se
incremento 100 veces durante el proceso agudo. Esto, indicó que la hepcidina se
comportaba también como un reactante positivo de fase aguda (1).
Gracias a la
Espectrometría de Masa, este péptido pudo ser
caracterizado como un polipéptido de 25 aminoácidos (Hpc-25) cuya estructura
espacial, lo define como una lámina plegada ß, con 8 residuos de cisteína cuyos
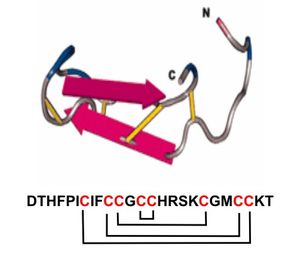
brazos están unidos por 4 puentes disulfuro(2). La Figura 1 muestra
una representación de la molécula de hepcidina activa.
Figura 1. Secuencia
aminoacídica y modelo tridimensional de la hepcidina humana. Notas:
Los terminales carboxilo y amino están indicados por C y N, respectivamente.
Los puentes de disulfuro se muestran en amarillo. En la secuencia primaria
mostrada debajo de la figura, se señalan estos puentes, que unen sus 8 residuos
de cisteína (3).
La Hepcidina es una hormona peptídica
producida principalmente en el hepatocito y que al igual que otras hormonas
peptídicas, es sintetizada inicialmente como un péptido de mayor peso molecular,
la pre-prohepcidina de 84 aminoácidos que luego es transformada en prohepcidina
de 60 aminoácidos y finalmente en su forma activa hormonal de 25 aminoácidos.
La región N-terminal de la prohormona es la que está involucrada en su función
reguladora, por lo que la pérdida de aminoácidos asociada con su transformación
de pre-prohepcidina a prohepcidina y finalmente a hepcidina, proviene del
extremo carboxilo terminal. Estas transformaciones se producen en los organelos
de los hepatocitos (retículo endoplasmático y trans-golgi) durante la síntesis
y distribución de la hepcidina. La enzima que se encarga de estas
transformaciones es una pre-proteína convertasa especial conocida como furina y
que se sintetiza al mismo tiempo que se sintetiza la hepcidina. Inhibidores de
esta enzima como la α-1 antitripsina afectan la conversión intracelular de pre-prohepcidina
a prohepcidina, así como la transformación de la prohepcidina a su forma activa
en el plasma. La eliminación de los 5 últimos aminoácidos de la región N terminal
resulta en la pérdida de su función (4,5,6).
Inicialmente se pensaba que la hepcidina
era sintetizada exclusivamente en el hígado, sin embargo investigaciones
posteriores han demostrado que el gen de la Hpc (HAMP) también se expresa en tejidos extrahepáticos. Kulaksiz
y colaboradores (2005) (7) demostraron que la Hpc también está
presente en las células apicales de los túbulos del riñón de humanos, ratas y
ratones, y postularon que esta hormona puede tener una función a nivel del
riñón y del tracto urinario. Luego, Schwarz y
colaboradores (2012) (8) evaluaron la expresión y localización de la
hepcidina gástrica en ratones y reportaron que la hepcidina se encuentra de
manera abundante en el fondo y en el cuerpo del estómago y que tanto la
Interleukina 6 como la infección por H.
pylori sobreestimulan su expresión. Estos mismos autores observaron que en humanos
la expresión de la hepcidina gástrica aumentó durante la infección por H. pylori y se normalizó después de una
exitosa erradicación y concluyeron que la hepcidina es un producto de las
células parietales, que son las mismas que regulan la acidez gástrica y que se
han asociado con el desarrollo de la úlcera gástrica bajo condiciones de estrés.
Más recientemente, se ha reportado que la hepcidina se produce en astrocitos y
microglías en el cerebro (9), posiblemente en la glándula mamaria (10),
así como en macrófagos y adipocitos (11). Sin embargo, la producción
hepática es la más importante y sirve
para satisfacer las necesidades de esta hormona tanto en el hígado como para
enviarla a otras células blanco (12), como son los enterocitos
duodenales, el propio hígado y los macrófagos del tejido retículo endotelial,
encargados de la eritrofagocitosis y reutilización del hierro de los
eritrocitos senescentes (11).
A continuación se discuten los aspectos más
importantes de la síntesis de la hepcidina, su efecto sobre su receptor
natural, la ferroportina y la función de la hepcidina-ferroportina sobre el
metabolismo y homeostasis del hierro.
Hepcidina y su papel
regulador en el metabolismo del hierro
Los primeros indicios sobre el papel
regulador de la hepcidina en el metabolismo del hierro aparecieron en los
estudios de Pigeon y colaboradores (2001)(13), quienes señalaron, que en ratones, la síntesis de la
hepcidina era inducida por el consumo de hierro y que este péptido se sobreexpresaba
ante una sobrecarga de hierro(13). Sin embargo, fueron los trabajos
de Nicolas y colaboradores (2001, 2002) con ratones transgénicos los que
mostraron el papel específico de la hepcidina sobre el metabolismo del hierro(14,15).
Estos investigadores observaron que en aquellos ratones donde se bloqueaba el
gen de la hepcidina, se desarrollaba una hemocromatosis con deposición de hierro
en hígado, páncreas y bazo (14), en contraste, en los ratones donde
se inducía una sobreexpresión del gen de la hepcidina, se desarrollaba una
anemia severa y la mayoría de las crías morían pocas horas después de nacer,
sugiriendo que la hepcidina inhibía la transferencia placentaria de hierro(15).
Estos estudios mostraron que la hepcidina es un péptido responsable de regular
tanto la captación de hierro a través del intestino como la liberación del
mismo por los macrófagos y los hepatocitos, el exceso de este péptido impide el
transporte de hierro a través del intestino y de la placenta y ocasiona una
carencia de hierro. En contraste, su deficiencia produce un aumento en la
absorción del hierro dietario, así como una sobrecarga del mismo en los
tejidos. Estos resultados llevaron a definir a la hepcidina como una hormona
que participa en la regulación del metabolismo del hierro. Esto se comprobó en
pacientes con hemocromatosis hereditaria (16).
Mecanismos de acción de la hepcidina como reguladora de la homeostasis de hierro
La identificación del mecanismo mediante
el cual la hepcidina regula el metabolismo del hierro se atribuye a los
trabajos de Nemeth y colaboradores (2004)(17) realizados en tejidos
celulares. Estos trabajos señalaron que la hepcidina impedía la salida de
hierro de la célula, debido a que esta hormona se unía al único exportador del
hierro celular, la ferroportina (FPN-1). La ferroportina es un péptido complejo,
que se une fuertemente a la membrana celular por medio de unos 9-12 dominios
transmembrana y que funciona como un monómero o un dímero. Este péptido es
esencial en eucariotes, para lograr el transporte de hierro entre células y
tejidos. Se han aislado tres tipos diferentes de ferroportina, pero en
mamíferos el único presente es el tipo FPN-1(18). Este
transportador se localiza en la superficie basolateral de los enterocitos del
duodeno, las células de Kupffer del hígado, y los macrófagos del bazo y la
médula ósea, así como en el tejido placentario(12), y su función es
exportar el hierro del interior de las células hacia la circulación. Con esto,
la ferroportina permite que tanto el hierro proveniente de la dieta
(enterocito) como el almacenado o reciclado dentro del organismo (hígado y
macrófagos) esté disponible para las distintas funciones corporales que lo
requieran. La inactivación del gen de la ferroportina impide la salida del
hierro del interior de las células y conduce a una acumulación del hierro
dentro de las células de Kupffer y de los macrófagos del bazo y a una
deficiencia sistémica del mismo(19).
Hasta la fecha, no se conoce en detalle
la forma en que FPN-1 transporta el hierro, pero se piensa que el sustrato es
Fe+2, ya que funciona en conjunto con una ferroxidasa multicobre
como la hefastina o la ceruloplasmina, que son enzimas capaces de reducir el
ion férrico. La presencia de estas ferroxidasas es esencial en el
funcionamiento de la FPN-1, ya que la carencia de esta actividad resulta en un acopio
de hierro en el transportador, su acumulación en la membrana o su
internalización y destrucción intracelular. La ferroportina, tal como ocurrió
con la hepcidina se descubrió en el año 2000(18) y desde entonces
se reconoce como el único receptor conocido para esta hormona reguladora del
metabolismo del hierro(11,18).
La hepcidina se une a la ferroportina en
uno de sus residuos de cisteína, que está ubicado en la posición C326 de su
estructura primaria. Esta unión entre FPN-1 y la hepcidina, estimula la unión
de la quinasa citoplasmática Janus-2 al complejo y la fosforilación de dos
tirosinas adyacentes, presentes en el lazo intracelular de la molécula de FPN-1.
El complejo [FPN-1-Hepcidina] ya fosforilado, estimula la formación de una
vesícula por invaginación de la membrana celular que lo encapsula y que ingresa
(internaliza) en la célula. En esta forma, llega al lumen de los cuerpos
multivesiculares y eventualmente el complejo se ubiquitiniza y se degrada por
la acción de enzimas lisosomales(18). Con esto, la hepcidina reduce
la concentración de FPN-1 en la membrana celular y en consecuencia, la
capacidad de la célula para exportar hierro. Por lo tanto, cambios en los
niveles de hepcidina circulantes, contribuyen a mantener los niveles séricos de
hierro dentro de los valores normales. Sin embargo, si la hepcidina aumenta o
disminuye fuera de lo normal, se producen cambios patológicos en la homeostasis
del hierro. Así, una hiperhepcidinemia, al ocasionar una excesiva degradación
de FPN-1, disminuye la absorción de hierro en el duodeno, restringe su salida
de los macrófagos, y reduce el hierro sérico. Con esto, baja la disponibilidad
del hierro para la síntesis de hemoglobina, produciendose deficiencia de hierro
y anemias. En contraste, una deficiencia de hepcidina aumenta la expresión de
FPN-1 sobre la membrana celular, con lo que se produce un aumento en la absorción,
así como en la salida del hierro de los macrófagos. De esta manera se excede la
capacidad de la transferrina para transportar hierro, con aparición de hierro
no unido a la transferrina que es captado por el hígado y otros tejidos,
produciendo morbilidades por exceso de hierro, como las observadas en los
pacientes con hemocromatosis (11). Actualmente, se investiga
activamente la acción de agonistas y antagonistas de la hepcidina que
potencialmente pudieran ayudar en el manejo de las anemias refractarias o en
los diversos tipos de hemocromatosis (12,20,21).
El efecto de la hepcidina en la
reducción de la expresión de FPN-1 en las células de Kupffer del hígado, y los
macrófagos del bazo, es más notable que en la células de los enterocitos
duodenales encargados de la absorción del hierro. Sin embargo, un incremento en
los niveles circulantes de hepcidina, resulta en una disminución importante en
la absorción del hierro (22). Esto se ha asociado con la observación
que en los enterocitos, la hepcidina no sólo produce la degradación de FPN-1
sino que, además reduce la síntesis del principal importador del hierro
dietario DMT1 (Transportador de metales divalentes) a través de un efecto del
complejo Hpc-FPN-1 en la transcripción del gen de DMT1(18).
En conclusión, más allá de los
mecanismos celulares que lo producen, un incremento de la hepcidina circulante
por encima de lo normal, al aumentar exageradamente la degradación de la
ferroportina y reducir su expresión en la membrana celular, disminuye la capacidad
de movilización del hierro de sus depósitos hepáticos, así como la
recirculación del hierro presente en los glóbulos rojos senescentes, incorporados
en los macrófagos principalmente del bazo. Asimismo, por sus efectos sobre el
epitelio intestinal, reduce la capacidad absortiva del hierro dietario. La
consecuencia de esto es una acumulación del hierro celular que conlleva a una
reducción del hierro circulante y de la capacidad eritropoyética, que eventualmente
produciría anemia. En contraste, una reducción exagerada en la hepcidina
circulante, causa una sobreexpresión de la ferroportina con lo que aumenta la
absorción y recirculación del hierro, así como sus niveles circulantes y de
depósito. Además se sobrepasa la capacidad de la transferrina con aparición de
hierro no unido a la transferrina en la circulación y su acumulación en el
hígado y otros tejidos. Todo esto eventualmente produce hemocromatosis y sus comorbilidades
asociadas (12).
Por estas razones, la función de la hepcidina en
el metabolismo del hierro se ha comparado con la función de la insulina en el
metabolismo de la glucosa, que en exceso puede producir episodios de
hipoglicemia y en déficit resulta en hiperglicemia y diabetes. En
consecuencia, la producción de hepcidina debe ser cuidadosamente regulada de
manera que permita mantener niveles adecuados de hierro para satisfacer las
necesidades metabólicas de este metal y al mismo tiempo evite el exceso y sus
efectos negativos (23).
Condiciones que afectan la síntesis de la Hepcidina
Entre
los factores que regulan la síntesis de hepcidina se encuentran la ingesta de
hierro, la anemia, la hipoxia y la inflamación. El hierro y la inflamación
estimulan la síntesis hepática de hepcidina, mientras que la anemia y la
hipoxia la inhiben (24,25,26,27). A continuación se discuten los
mecanismos que regulan la síntesis de hepcidina en cada una de estas
condiciones.
Producción de hepcidina
en condiciones de alto hierro
La síntesis de la hepcidina está
regulada de manera transcripcional y hasta la fecha no hay evidencias de otro
tipo de control. Los modelos con ratones transgénicos han aportado una valiosa
información sobre los mecanismo moleculares que participan en la regulación de
la transcripción del gen de la hepcidina, conocido como gen del péptido
antimicrobiano hepcidina (HAMP) y que
es inducida por el hierro, a través de la vía de señalización celular conocida
como vía SMAD. En esta vía están involucradas una serie de proteínas, siendo el
centro de esta cascada de señalización la Proteína Morfogenética del Hueso (BMP6)
y su receptor celular rBMP (27).
Es
importante señalar que las proteínas BMP son al menos 8 péptidos diferentes y
constituyen una subfamilia dentro de la superfamilia de proteínas
multifuncionales conocidas como, Factores de Crecimiento Transformantes tipo ß
(TGF-ß). Esta superfamilia incluye más de 60 proteínas, que participan en una
gran gama de funciones como son diferenciación y proliferación celular, inhibidores
del crecimiento, modulación del sistema inmune e inflamación, cicatrización de
tejidos, reproducción, producción de matriz extracelular y formación de hueso,
desarrollo embrionario, neurogénesis, etc. (28). De los diferentes
tipos de proteínas morfogenéticas del hueso, sólo la BMP6 se reconoce como la
que realmente tiene una función regulatoria en el metabolismo del hierro y en
la transcripción del gen de la hepcidina (11). Algo que es común a
todas las proteínas multifuncionales que pertenecen a la superfamilia (TGF-ß) es
que, cuando alguno de sus miembros, presentes en el torrente sanguíneo, se une
a sus receptores específicos en la membrana celular, usan una vía de
señalización celular única, que es la vía conocida como vía SMAD.
La vía
SMAD está formada por una serie de proteínas SMAD
que están presentes en el citoplasma celular en forma de monómeros inactivos.
Sin embargo, cuando alguno de los miembros de la familia (TGF-ß) se une
a su receptor celular, las proteínas SMAD se fosforilan en sus residuos serina
o treonina y con esto, cambian su conformación y se ensamblan entre sí para
formar complejos (homodímeros o trímeros o heterodimeros o heterotrímeros) que
pueden atravesar la membrana nuclear y unirse a elementos de unión SMAD
presentes en la molécula de DNA, con lo que se estimula o desestimula la
transcripción de un gen específico. Este es un proceso complejo, en el que no
sólo participan las proteínas SMAD sino también otras proteínas y factores de
transcripción. Así, la gran variedad de ligandos (componentes de la superfamilia
TGF-ß), de proteínas SMAD y sus complejos, y las demás proteínas y factores de
transcripción participantes, hacen que la vía de señalización SMAD pueda actuar
en un gran número de procesos celulares, entre ellos, la síntesis de hepcidina,
dependiente de una alta concentración de hierro.
En
el caso de la síntesis de la hepcidina, participan el BMP6 que se considera el
ligando especifico para la síntesis de esta hormona y las proteínas SMAD 1,5 y
8. Además, en la activación de esta vía de señalización, participa el SMAD 4 que
también se conoce como co-SMAD o SMAD constitutivo, ya que es común a todas las
vías de señalización SMAD (6,27,28).
Con base a estudios realizados por muchos
grupos de trabajo, se ha construido un modelo para explicar el mecanismo
molecular que regula la expresión de la hepcidina por el hierro. En este
modelo, el mecanismo de regulación y control recae sobre el receptor del BMP6 y
otros componentes que participan en la activación de la vía de
señalización SMAD. Esto se muestra en la
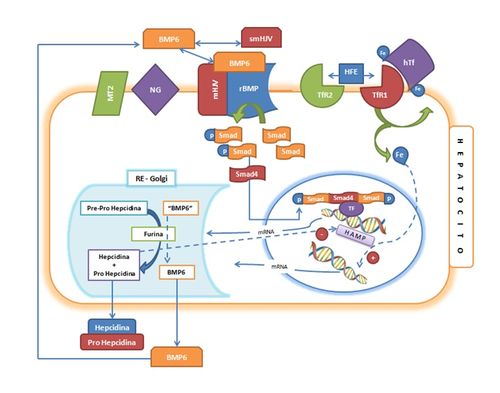
Figura 2, que resume lo reportado por varios autores (6, 11,12, 21,27).
Figura
2. Modelo
de los mecanismo moleculares que participan en la regulación de la expresión de
la hepcidina por el hierro. Abreviaturas: BMP6: Proteína Morfogenética del Hueso, tipo6 y miembro del grupo de
Factores de Crecimiento Transformantes de
la familia TGF-ß. BMP6: Precursor de
BMP6 de más alto peso molecular. MT2: Serino proteasa tipo 2 o
Matriptasa 2. NG: Neogenina. mHJV:
Hemojuvelina de membrana, coreceptor de BMP6. smHJV: Hemojuvelina soluble. hTf:
Holo transferrina o transferrina diférrica. TfR1: Receptor de
transferrina tipo1. TfR2:
Receptor de transferrina tipo 2. HFE:
Proteína de susceptibilidad a la hemocromatosis tipo I. rBMP: Receptor de BMP6. SMAD:
Proteínas transductoras de las vías de señalización del BMP. SMAD4: SMAD constitutivo, común a todas
las vías de señalización SMAD. TF:
factores de transcripción que participan en la unión del complejo Smad con las
regiones de respuesta del gen de la hepcidina (HAMP). mRNA: RNA mensajero. RE-Golgi:
Reticulo endoplasmático-Aparato de Golgi. Furina:
proproteína convertasa (endopeptidasa) involucrada en la conversión de los
precursores de la hepcidina y Factores de Crecimiento Transformantes de la
familia TGF-ß en sus formas activas.
La Figura 2 muestra una célula hepática que expresa un
receptor para BMP6 (rBMP) sobre la membrana celular. Este receptor esta unido a
una molécula de hemojuvelina de membrana (mHJV), que es una proteína que actúa
como un co-receptor y que se une al ligando (BMP6). Sólo en esta forma (BMP6-mHJV)
el BMP6 puede interactuar con su receptor. La unión del BMP6 con su receptor,
permite la fosforilación de los factores de señalización SMAD que cambia su
conformación y les permite interactuar con el SMAD constitutivo (SMAD4) y
formar complejos que migran hacia el núcleo. En el núcleo este complejo
(heterotrímero), en conjunto con otras proteínas y factores de transcripción(28)
interactúa con el DNA, en sitios de respuesta específicos, ubicados en el gen
promotor de la hepcidina (HAMP), estimulando su transcripción. Con esto, se inicia
la transcripción de HAMP con la generación de un mRNA que codifica para la
molécula de Pre-prohepcidina (Fig. 2).
Otros sensores que participan en esta vía de señalización
(Fig. 2) son las concentraciones de transferrina diférrica u holotransferrina (hTf),
las concentraciones de los receptores de transferrina TfR1 y TfR2 y su
contraparte transmembrana, la proteína de sensibilidad a la hemocromatosis
hereditaria tipo 1 (HFE).
El incremento de las concentraciones de transferrina
diférrica (hTf), cambia la afinidad de
HFE por el receptor de transferina 1 (TfR1) hacia el receptor de transferrina 2
(TfR2). Con esto, la transferrina diférrica (hTf) unida a TfR1, se internaliza
en el citoplasma celular por endocitosis, libera el hierro dentro de la célula para
su metabolismo o para incrementar los depósito de hierro en las moléculas de
ferritina, mientras el complejo TfR1- Tf
libre de hierro, se recicla a la membrana y la apotransferrina (Tf)
vuelve al suero para captar más hierro (23). Al mismo tiempo, el
complejo HFE-TfR2, estimula la interacción de BMP6 con la hemojuvelina de
membrana (mHJV) activando la vía de señalización BMP-SMAD favoreciendo así, la
transcripción del gen de la hepcidina (HAMP) (6,11,29) (Fig 2). Con esto, un incremento en la
concentración sérica de hierro resulta en la producción de un mRNA que codifica
para la hepcidina y además, en un aumento en la concentración de hierro celular
que resulta en la generación de un mRNA que codifica para BMP6. Una vez
completado el proceso de transcripción de estas moléculas de mRNA, ellas migran
al citoplasma celular para su traducción, modificación postransduccional y
extrusión al espacio extracelular en el sistema retículo endotelial y el
aparato de Golgi (RE-Golgi) (11,30,31).
Algunos autores han señalado que el hierro también puede
inducir la síntesis de BMP6 en el intestino y que en este caso, el BMP6
sintetizado, migra al hígado donde también participa en la regulación de la
producción de hepcidina (32).
Modificaciones postraduccionales de la molécula de hepcidina
De la discusión anterior (Fig. 2), se desprende que
una sobrecarga de hierro estimula la transcripción del gen HAMP de la
hepcidina y del gen de la proteína multifuncional BMP6, que es el ligando que
activa la vía de señalización BMP6-SMAD. Esto significa que en estas
circunstancias, la secuencia de bases de estos genes se transcribe en moléculas
de RNA mensajero, que migran del núcleo al sistema ribosomal. Allí, sirve de
guía para sintetizar la secuencia primaria de la hepcidina y BMP6.
En el caso de la hepcidina, la cadena polipeptídica
sintetizada durante este proceso tiene 84 aminoácidos y contiene dos péptidos
adicionales a los presentes en la hepcidina activa, que tiene sólo 25
aminoácidos (hepcidina-25). A la cadena de 84 aminoácidos se le conoce como
pre-prohepcidina y contiene un péptido señal de 24 aminoácidos que se pierde
durante la migración del polipéptido desde el retículo endoplasmático al trans-golgi
para generar prohepcidina. Luego pierde un segundo péptido de 35 aminoácidos
con lo que produce la hepcidina madura o activa. La conversión de pre-prohepcidina
a prohepcidina y finalmente a la hepcidina activa, la cataliza una proproteína convertasa conocida como furina
que es una endopeptidasa que corta secuencialmente estos péptidos
posteriormente a la síntesis de la estructura primaria (4,5,6).
La furina es una enzima presente en la membrana del sistema trans-Golgi (Fig.
2) en los endosomas y la membrana plasmática y que está presente en todos los
tejidos y contribuye en el proceso de secreción y maduración de muchas
proteínas fisiológicamente activas (33). Este elaborado proceso de
síntesis es frecuente en proteínas funcionales que requieren de un plegamiento
determinado que les permite lograr la estructura tridimensional apropiada a su
función, así como facilitar el proceso de secreción desde las células que las
producen. Otros péptidos fisiológicamente activos también se sintetizan de esta
forma, esto ocurre en el caso de la insulina en las células ß del páncreas(34),
la paratohormona en la
paratiroides(35) y en el caso
que nos ocupa, tanto los Factores de Crecimiento Transformantes de la familia
de los Tgf-ß a los que pertenece el BMP6(28) y la hepcidina en las
células hepáticas(6). En estos tres últimos casos, la proproteína
convertasa que participa en los procesos post-transduccionales que ocurren
durante su formación y activación es la furina. La discusión anterior señala que
la furina juega un papel muy importante en la síntesis de la hepcidina, ya que
modifica y activa al péptido precursor de la hepcidina madura. Sin embargo,
además de esta función post-traduccional, la furina también afecta la
transcripción del gen de la hepcidin (HAMP). Esto ocurre ya que esta enzima, a
través de un proceso de proteólisis limitada, elimina el péptido de unión de la
Hemojuvelina (mHJV) con la membrana celular, de manera que esta se desprende de
la célula y se libera en forma soluble (sHJV) en el medio extracelular (Fig. 2).
Esto es esencial, ya que sólo la Hemojuvelina de membrana (mHJV) es la que
puede presentar a la molécula de BMP6 a su receptor. Además, la Hemojuvelina
soluble, en el suero, mantiene su afinidad por BMP6, limitando así, su interacción
con la mHJV. Con esto, se reduce su capacidad de presentación del BMP6 a su
receptor celular que es el iniciador de la transcripción del gen de la
hepcidina (6). En esta acción, otro componente del sistema de
señalización de la transcripción del gen de la hepcidina (HAMP), la Neogenina
(NG) (Fig. 2) participaría en el traslado de la mHJV al trans-golgi para que la
furina pueda ejercer su función y generar
sHJV (6).
La conversión de pre-prohepcidina a prohepcidina y
finalmente a hepcidina madura, no es completa. Esto, se atribuye a la presencia
de inhibidores de esta enzima como es el α-1 antitripsina (5). La
consecuencia de esta inhibición es que a nivel celular se mantiene una
concentración de prohepcidina que puede unirse al gen promotor de la hepcina y
reducir su transcripción. De esta manera, la prohepcidina actuaría como un
regulador de la expresión de la hepcidina, dependiente de la actividad de la furina
y sus inhibidores. Otra consecuencia de esta inhibición es que en el suero
circulan tanto la hepcidina como la prohepcidina (Fig. 2) en concentraciones
más o menos equivalentes (36). En el suero, la hepcidina circula preferentemente unida a la α-2-macroglobulina
pero también circula libre o ligada a la albúmina (37), mientras que
la prohepcidina circula unida a la molécula de α-1 antitripsina (5).
Otra observación importante en relación con la
furina es que su producción es dependiente de los niveles de hierro y utiliza
para activar su síntesis, rutas similares a las que activan la producción de
hepcidina, en condiciones de alto hierro. El mecanismo no está aun resuelto,
pero se sabe, que un actor importante a nivel de membrana es el complejo
TfR2-HFE, que se forma por desplazamiento de HFE del TfR1 cuando este interactúa
con la holotranferrina proveniente del suero (Fig. 2). Así, en condiciones de
alto hierro sérico, la tranferrina le entrega el Fe a TfR1, que se internaliza
y recicla después de entregar su hierro y con esto, se desplaza HFE desde TfR1
a TfR2 y este complejo (TfR2-HFE) por una parte activa al receptor de BMP y también
estimula la transcripción de la furina (6).
En este complejo sistema de señalización, también
actúan otras proteasas. Una de estas es la Matriptasa-2 (MT2) (Fig. 2) que como
la furina, es una endoproteasa. La MT2, también actúa sobre la mHJV, pero el
producto es una HJV inerte que no tiene afinidad por BMP6, por lo que en
condiciones en que esta enzima esté activada, se reduce la producción de
hepcidina (21). Se ha sugerido que la Matriptasa-2 es importante en
la regulación de la producción de hepcidina en condiciones de bajo hierro (11),
ya que su actividad se induce al disminuir el hierro celular, probablemente por
sobreexpresión del gen TMPRSS6 que es el que la codifica. Con esto, disminuye
la mHVJ y en consecuencia su capacidad para presentar el BMP6 a su receptor y
así también se reduce la producción de hepcidina.
Es importante señalar que la identificación de los
componentes que participan en la transcripción del gen HAMP y su respuesta al
estado del hierro, se ha logrado gracias al estudio de sus efectos en
enfermedades hereditarias en humanos que afectan al propio gen HAMP, a
mutaciones en el gen de la HJV, en el de la Matriptasa-2, en el de la proteína
de la hemocromatosis hereditaria, HFE, en el de receptores de transferrina tipo
2 (TRF2) etc. Adicionalmente, la utilización de ratones o células de ratones
con genes noqueados o silenciados por medio de la ingeniería genética sometidos
a dietas ricas y deficientes en hierro, también han sido muy útiles en la
identificación y funcionamiento del sistema de transcripción del gen HAMP y la
producción de hepcidina. En todos estos
casos, se reduce la transcripción y síntesis de hepcidina, con lo que se pierde
la capacidad de control sobre la ferroportina. En consecuencia, aumenta
indiscriminadamente la absorción de hierro y con esto, el riesgo de producir
una acumulación de hierro y hemocromatosis, tal como ocurre en las enfermedades
hereditarias ya mencionadas.
Desde un punto de vista conceptual, el descubrimiento de la hepcidina y
su función regulatoria de la extrusión del hierro celular, por su acción sobre
la ferroportina en todas las células y además sobre la expresión de DMT1 y sus
efectos sobre la incorporación del hierro dietario desde el lumen del intestino
a las células de la mucosa, han representado un avance notable en el
conocimiento de la función de la mucosa intestinal en el mantenimiento de la
homeostasis del hierro. Al respecto, es importante recordar que hasta finales
del siglo XX y comienzos del XXI, el concepto predominante era que la mucosa
intestinal era capaz de detectar el estado nutricional del hierro y regulaba la
absorción de este metal de acuerdo con las necesidades del organismo. Así, el
hierro presente en las células epiteliales, podía ingresar al organismo, de
acuerdo con sus necesidades o permanecer en estas células, asociadas con las
moléculas de ferritina para perderse en las heces en el proceso de exfoliación
de las células en la cumbre de las vellosidades. De acuerdo con esto, los
individuos deficientes en hierro, incorporarían menos hierro en la ferritina y
en consecuencia absorberían más (38). Sin embargo, el mecanismo por
el cual las células de la mucosa percibían el estado del hierro no se conocía. En
la actualidad con el descubrimiento de la hepcidina este concepto ha cambiado (23).
Ahora sabemos que el hígado es el órgano sensor tanto de los niveles
circulantes como de los depósitos de hierro (11) y que produce la
hormona hepcidina en mayor o menor cantidad cuando estos niveles aumentan o
disminuyen, respectivamente. Luego es la hepcidina la que actúa sobre la mucosa
para que aumente o disminuya la absorción del hierro, manteniendo una
homeostasis apropiada, a pesar de las variaciones en el consumo o en las
pérdidas de hierro.
Regulación de la síntesis de Hepcidina por la inflamación
La inflamación tiene un efecto muy potente sobre la
homeostasis del hierro. En presencia de inflamación, disminuye la absorción
intestinal de hierro, hay un secuestramiento del mismo por el hígado y los
macrófagos, y como consecuencia de esto, se produce una disminución del hierro
circulante que cursa con depósitos de hierro normales o elevados y que eventualmente
termina en la anemia de las enfermedades crónicas, tales como enfermedades
crónicas del riñón, infecciones prolongadas, diabetes, trauma severo, artritis
reumatoidea, enfermedad de Chron y otras enfermedades inflamatorias del aparato
gastrointestinal, cáncer, etc. En general, patologías asociadas con un estímulo
crónico del sistema inmune. Actualmente, es ampliamente aceptado que la anemia
asociada con las enfermedades crónicas es el resultado de una estimulación de
la producción de hepcidina(6,12,21,27). Esto se debe a que el
promotor del gen de la hepcidina (HAMP) no sólo posee elementos de respuesta a oligómeros
SMAD (sensibles al hierro) como ya se discutió, sino también a dímeros STAT
(sensibles a la inflamación) (6,11,12,21).
Experimentos en cultivos de células hepáticas
humanas han mostrado que la IL-6, así como otras interleucinas inflamatorias y
los lipopolisacáridos presentes en las membranas bacterianas, inducen la
expresión de hepcidina. Por otra parte, en voluntarios humanos los niveles de
hepcidina urinaria se incrementaron hasta 7 veces pocas horas después de una
infusión de lipopolisacáridos o de IL-6 (27), señalando que la
hepcidina funciona como un reactante de fase aguda. Esto, está de acuerdo con
las primeras observaciones relacionadas con este péptido al que se le asignó
una función antimicrobiana. Así, antes de conocer su función reguladora del
metabolismo del hierro, a la hepcidina se le conocía como LEAP-1 (Liver
expressed antimicrobial peptide1) (39). En relación con esto, la
visión actual es que la hepcidina funciona como una defensina que ejerce su
actividad antimicrobiana mediante deprivación de hierro, que es un elemento
crítico para la proliferación bacteriana (6). Esto se sustenta en hallazgos
que datan de los años 70 y que indican que las infecciones se vuelven más
severas en condiciones de hiperferremia, la suplementación con hierro en
condiciones de infección aumenta la severidad de la infección y comprometen la
vida del paciente (40,41). Durante la infección y trauma el organismo
tiene mecanismos para secuestrar el hierro en los depósitos y reducir el hierro
sérico (42), disminuyendo así su disponibilidad para los
microorganismos invasores (43). Este efecto es mediado por un
aumento en la síntesis de hepcidina y sus niveles circulantes.
El efecto estimulante de la IL-6, así como el de
otras citoquinas como IL-1, IL-2, IL-7, IL-12, IL-17, IL-23 o mediadores de
inflamación como lipopolisacaridos (LPS), oncostatina, turpentina, etc. sobre
la hepcidina es transcripcional y dependiente de la vía de señalización STAT3
(Fig. 3).
Estos agentes inflamatorios se unen a sus
receptores de membrana y producen la fosforilación de los activadores de
transcripción STAT3. Esta fosforilación esta mediada por la kinasa Janus, JAK2.
La actividad JAK2/STAT3 promueve la dimerización de los activadores STAT3 y su
migración al núcleo celular, donde actúan como factores de transcripción nucleares, que tienen afinidad por la sección
del DNA donde se ubica el gen promotor
de la transcripción de la hepcidina (HAMP) para producir hepcidina (6,11,12,21,27).
Es importante indicar que aunque el efecto de la IL-6 ha sido el más estudiado,
el efecto estimulante de los LPS en la producción de hepcidina se ha detectado
incluso en animales genéticamente modificados para anular la producción de esta
interleuquina. También hay que señalar que la estimulación de la producción de
hepcidina por la vía inflamatoria recién descrita requiere de un sistema
BMP/SMAD intacto, ya que en animales en que este sistema está alterado, no se
produce el aumento en la producción de hepcidina asociado con la inflamación (11,21,45).
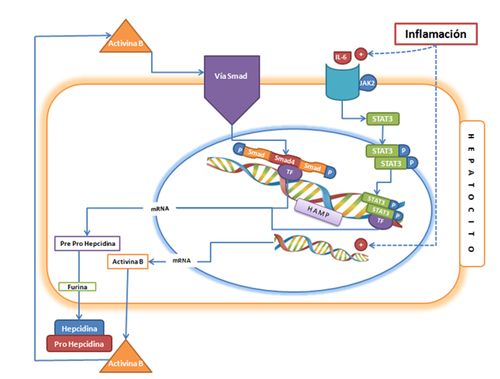
Figura 3. Modelo
de los mecanismos moleculares que participan en la regulación de la expresión de
la hepcidina por la inflamación (6,11,12,21,27,44). Notas: IL-6:
Citoquina proinflamatoria. JAK2:
Tirosina quinasa 2. STAT3: polipéptido
involucrado en la vía de señalización celular STAT capaz de activar el gen HAMP
en respuesta a la inflamación. Activina
B: Proteína multifuncional de la misma familia que BMP6 que se produce en
respuesta a la inflamación. Para más detalles ver Figura 2.
Adicionalmente, se ha demostrado que otros Factores
de Crecimiento Transformantes de la familia TGf-ß, específicamente las Activina
A y B, que usualmente se han asociado con la regulación de los procesos
reproductivos, aumentan su expresión en respuesta a enfermedades inflamatorias
tales como septicemia, enfermedades inflamatorias del tracto gastrointestinal y
artritis reumatoidea (44) y que estimulan la vía SMAD, aumentando la
expresión de la hepcidina. Esto se detectó en el hígado de ratones tratados con
LPS, en los que se observó un incremento en la fosforilación de los SMAD 1/5/8
y producción de precursores de la hepcidina que fue independiente de BMP6 y Fe.
Estas observaciones evidencian un papel no sólo para la vía STAT-3, sino
también de la vía SMAD en la inducción de la producción de hepcidina en
respuesta a la inflamación (44) y explican por qué en condiciones
inflamatorias en que no aumenta la IL-6, puede incrementarse la producción de
hepcidina. Estos conceptos se integran en la Figura 3 que muestra como en un
hepatocito, la inflamación por acción de la IL-6 y su receptor de membrana,
activa la vía de señalización JAK-STAT y al mismo tiempo estimulan la
producción de Activinas, que estimulan la vía de señalización SMAD. Estos estímulos,
actuando por vías de señalización diferentes pueden incrementar la producción de
hepcidina, afectando negativamente la absorción del hierro dietario, así como
la salida del hierro celular (hígado, macrófagos) por la acción de la hepcidina
sobre la ferroportina y DMT1. Con esto se produce un secuestramiento del hierro
con disminución del hierro sérico y eventualmente la anemia secundaria a la
inflamación que caracteriza al enfermo crónico (46).
Regulación de la síntesis de Hepcidina por hipoxia y su relación con la eritropoyesis
La hipoxia aumenta la formación de radicales libres
de oxigeno (ROS) y estos reducen la activación transcripcional de HAMP. Este
efecto de los ROS, puede prevenirse con el uso de antioxidantes. Se postula que
los ROS bloquean el acceso de los factores de transcripción (STAT ó SMAD) al
gen promotor de HAMP, con lo que desactivan la expresión de la hepcidina (6).
Este tipo de hipoxia se asocia con condiciones agudas que afectan a un tejido o
tejidos particulares, como ocurre en casos de la isquemia-reperfusión que
resulta de las cirugías cardíacas o de otros tejidos, del ejercicio intenso que
provoca hipoxias musculares, de las hipoxias neonatales o traumas que afectan al
pulmón y las vías aéreas. Sin embargo, hay hipoxias de tipo crónico como las
asociadas con problemas bronco pulmonares, insuficiencias cardíacas o anemias
en las que el organismo intenta compensar la falta de oxigeno, con un aumento
de la eritropoyesis. Una situación similar se ve en casos de hemorragias o hemólisis,
así como en anemias como la falciforme y la talasemia en las que se producen
glóbulos rojos provistos con una hemoglobina limitada en su función de
transportar oxigeno (47). En todos estos casos en que aumenta la
eritropoyesis, independientemente de la causa y tal como ocurre en las anoxias
agudas, la producción de hepcidina disminuye, aunque los depósitos de hierro
sean normales o elevados (6,12,21).
Los factores específicos responsables de la
reducción en la producción de hepcidina, tanto en casos de hipoxias agudas o en
situaciones en las que una elevada eritropoyesis intenta compensar la hipoxia,
aun no se conocen. Uno de los posibles, es el factor inducible por la hipoxia
(HIF) que es un factor de transcripción presente en todas las células y que
actúa sobre la expresión de cientos de genes en casos de hipoxia y que afecta al
metabolismo del hierro estimulando la producción de eritropoyetina en el riñón,
así como la producción de transferrina y sus receptores (48). Este
factor también estimula la producción de la enzima xantina oxidoreductasa que
funciona en la degradación de las purinas, pero que en condiciones de hipoxia
actúa como una oxidasa (xantina oxidasa), produce superoxido o peróxido de
hidrógeno y puede además liberar el hierro de la ferritina y generar ROS
utilizando las reacciones de Fenton (49). En estos casos, tanto la
generación de radicales libres como la hipoxia, inhiben la producción de
hepcidina.
Una observación importante es que, para que se
reduzca la producción de hepcidina en los casos de anemias con eritropoyesis
aumentada es imprescindible tener una médula ósea intacta. Por esta razón, se
piensa que la médula secreta un factor eritroide que reduce la producción de
hepcidina en proporción a la actividad eritropoyética y su producción podría
depender de la eritropoyetina (11,12,21).
El hecho que la anoxia con eritropoyesis aumentada,
reduzca la síntesis y niveles séricos de la hepcidina, puede complicar la
interpretación de los niveles séricos de hepcidina en los casos de las anemias
de los enfermos crónicos, ya que en ellos la inflamación estimula, mientras que
la anemia inhibe la producción de hepcidina (12).
La reducción de la hepcidina en condiciones de
anoxia y eritropoyesis aumentada, produce los efectos contrarios a los que
ocasiona un aumento de la hepcidina. Así, una disminución de la hepcidina deja
sin control los niveles de ferroportina en las células del intestino,
macrófagos del bazo y células parenquimatosas del hígado. Con esto aumenta la
absorción del hierro, y su liberación de los depósitos hepáticos y de los
macrófagos del bazo. En consecuencia, aumentan los niveles de hierro circulante,
así como el porcentaje de saturación de la transferrina. Estos efectos de la
hepcidina sobre el metabolismo del hierro, son convenientes, ya que en
conjunción con un aumento en los receptores de tranferrina en las células
eritropoyéticas, aumenta la producción de glóbulos rojos y con ello la
capacidad de transportar oxigeno y compensar la anemia ferropénica y la
consecuente anoxia (23). Sin embargo, una situación diferente se
plantea en el caso de anemias no dependientes del hierro sino en defectos
genéticos de la hemoglobina, como son la anemia falciforme o la talasemia o de
enzimas específicas esenciales en el metabolismo y protección de los
eritrocitos, tal como ocurre en los pacientes con defectos genéticos de la
enzima glucosa-6-fosfato deshidrogenasa. En estos casos la anemia (hemolítica)
y la anoxia asociada persisten a pesar del aumento en la disponibilidad del
hierro producido por la baja hepcidina. La consecuencia de esto es un aumento
en el hierro sérico y de hierro no ligado a la tranferrina ,con acumulación de hierro en el bazo y el hígado que
eventualmente produce complicaciones que pueden ser letales (47,50).
Funciones de la prohepcidina
Así como la hepcidina tiene un papel muy
bien definido en la homeostasis del hierro, la situación no es la misma para la
prohepcidina. Este péptido, sin duda tiene un papel como precursor de la
hepcidina ya que intracelularmente, por acción de la furina genera hepcidina (6,11,51,52).
Además, la prohepcidina en el núcleo, se puede unir al gen de la hepcidina
inhibiendo su expresión (36). Sin embargo, estas funciones las
cumple a nivel celular, pero no está clara su función en el fluido
extracelular. Es interesante que en el suero las concentraciones de hepcidina y
prohepcidina sean similares, lo que sugiere que debería tener una función. Es
posible, que tal como ocurre en el caso de otras hormonas como las tiroideas o
el calcitriol, e incluso en el de los Factores de Crecimiento Transformante
(TGF-ß), la prohormona sérica sirva como una fuente de la hormona activa que se
transforma a esta cuando sea necesario. Sin embargo, esto no se ha establecido
en el caso de la prohepcidina. Sólo se conoce que la prohepcidina no tiene
efecto sobre la ferroportina a menos que se transforme en hepcidina activa
(Hpc-25) por acción de la furina (51).
Se dispone de una abundante literatura
que relaciona los niveles de prohepcidina con la incidencia de anemias en
varios tipos de pacientes. Por ejemplo, en problemas asociados con el sistema
gastrointestinal (53,54), con obesidad (55), con mieloma
múltiple(56), con anemias de enfermos crónicos(57), con
enfermedades renales(58,59), etc. En estos estudios, se hacen comparaciones
de los niveles promedio de prohepcidina entre tratamientos y además se estudia
la correlación de los niveles de esta prohormona con los indicadores del estado
nutricional del hierro y de los indicadores de inflamación típicos de cada
patología. En general, estos trabajos muestran que los valores de prohepcidina
resultan similares entre tratamientos, pero correlacionan significativamente ya
sea con algunos de los indicadores de hierro o de la inflamación o ambos y la
mayoría de los autores señalan que la prohormona cumple una función, aun no
definida, en el metabolismo del hierro.
La hepcidina se determina en su forma más
rigurosa usando un sistema acoplado de cromatografía y espectrometría de masa.
Este método logra discriminar bien a la hepcidina de 25 aminoácidos de la
prohepcidina de 60 aminoácidos(60). Sin embargo, para aplicar esta
metodología se requiere de equipos sofisticados que habitualmente no están
disponibles en laboratorios comunes. Por esta razón, se utilizan métodos
basados en anticuerpos contra estos péptidos. Estos métodos en general correlacionan
bien con la espectrometría de masa y su utilización ha incrementado
notablemente la investigación en esta área (21). El primero de estos
métodos que se desarrolló fue el de la prohepcidina, ya que es más fácil
producir anticuerpos contra un péptido de 60 aminoácidos que contra uno de sólo
25. Luego, aproximadamente en el año 2010 se comenzó a comercializar un kit
para la determinación de la hepcidina-25 y este es el que se utiliza con mayor
frecuencia en la actualidad. Además, la
facilidad de contar con métodos apropiados para determinar tanto la hormona
como la prohormona ha permitido establecer comparaciones entre ellas.
Analizando esta información (61,62,63), se observa que los niveles
séricos de la prohecidina y la hepcidina son similares, que los coeficientes de
correlación entre ellas no son muy altos y tienen signo negativo, indicando que
cuando una de ellas aumenta, la otra disminuye. Ambos péptidos correlacionan
significativamente con las medidas del estado nutricional del hierro o la
inflamación. Los autores de estos trabajos Oustmanolakis P y colaboradores (2010
y 2013)(62,63), señalan que tanto la prohepcidina como la hepcidina
juegan un papel importante en la etiología de las anemias asociadas con las
enfermedades inflamatorias del aparato gastrointestinal (IBID) y sugieren que
debe existir una vía común entre estos péptidos que determine el balance entre
ellos. Todos los autores recomiendan continuar estudiando ambos péptidos en el
caso de IBID.
Integración de la regulación del metabolismo del hierro
En los escasos 12 años que han pasado
desde las primeras publicaciones sobre la hepcidina, se ha destacado la
importancia central de esta hormona en el metabolismo del hierro y en la
patogénesis de los diferentes desordenes asociados con el hierro, como son los
casos de la hemocromatosis o las anemias refractarias al tratamiento con hierro(12). Sin embargo, no se pueden ignorar los demás mecanismos
implicados en esta regulación. Entre ellos, la función de la eritropoyetina que
es fundamental en la actividad del tejido eritropoyético, tanto en la síntesis
de nuevos glóbulos rojos y hemoglobina en condiciones de anoxia y que se
estableció entre los años 40 y 50 (64), así como la función de las
proteínas sensibles al hierro (IRP1 y IRP2), que descubiertas en base a los
trabajos pioneros de Hamish Munro en el MIT (65) y muchos otros (66),
mostraron que en condiciones de bajo hierro, IRP1 o IRP2 tienen la capacidad de
unirse con alta afinidad a sitios de respuesta de los RNA mensajeros (IRE) de
la ferritina (IRE-IRP en la región 5´), o de los receptores de transferrina (IRE-IRP
en la región 3´). El efecto de esta unión, en el caso de la ferritina, es inhibir
la síntesis de sus cadenas H y L, mientras que en el caso de los receptores, inhibir
la degradación del mRNA que codifica su estructura, estabilizándolo y así
aumentando la capacidad de síntesis de las proteínas que los conforman. Estos
hallazgos del siglo XX se utilizaron para explicar los cambios en el
metabolismo y homeostasis del hierro hasta finales de los años 90 e inicios de
los años 2000. Actualmente se sabe que una de estas proteínas (IRP1) es la
aconitasa citosólica(67), que en condiciones de bajo hierro pierde uno
de sus centros de hierro-azufre, deja de funcionar en el ciclo de los ácidos
tricarboxílicos y así, en el metabolismo de los carbohidratos, para
transformarse en un sensor de los niveles de hierro. IRP2 cumple las mismas
funciones que IRP1, también pertenece a la familia de las aconitasas, pero por
tener sólo 3 centros hierro-azufre no participa en el ciclo de Krebs. Sin
embargo, su síntesis aumenta en condiciones de bajo hierro, mientras que cuando
el hierro aumenta, se degrada perdiéndose así su efecto regulatorio sobre la
síntesis de ferritina o de los receptores de transferrina(67).
Es importante señalar, que no sólo la
síntesis de la ferritina y los receptores de transferrina están sujetos a la
regulación por proteínas sensibles al hierro (IRP), sino que este mecanismo de
regulación dependiente de hierro también afecta a otras proteínas(23, 66).
Así, la síntesis del transportador de metales divalentes (DMT1) aumenta en
casos de bajo hierro por tener zonas IRE-IRP en la región 3´ de su mRNA,
mientras que en la misma situación, la síntesis de la ferroportina y la de la
enzima reguladora de la síntesis de hemoglobina presente en los reticulocitos y
tejido hematopoyético (δ-Amino Levulinato Sintasa-2) disminuyen por tener zonas
IRE-IRP en la región 5´de sus mRNA. Sin embargo, en estos casos, por razones
aún en estudio, el efecto del hierro es menor(66).
Este tipo de regulación, se utilizó para
explicar algunas características muy importantes de la homeostasis del hierro
como es el caso de las variaciones en la absorción de hierro en respuesta al
estado nutricional del hierro.
Una característica del hierro, que lo
diferencia de los demás nutrientes es que el contenido corporal del hierro es
altamente conservado y en ausencia de sangramientos o aumento en los
requerimientos como ocurre durante el embarazo, crecimiento o la lactancia, las
pérdidas de hierro son muy pequeñas, prácticamente constantes (0.98 a 2.4
mg/día) (68) y no están sujetas a ningún tipo de regulación (69).
Debido a esta falta de regulación en las pérdidas, el hierro que debe reponerse
para compensarlas depende exclusivamente de variaciones en la cantidad neta de
hierro absorbida y que de acuerdo con el viejo paradigma, se atribuía a las
células de la mucosa intestinal (70). Un componente fundamental en esta
modulación se atribuía a la molécula de ferritina, conocida desde 1937 (71)
como una proteína capaz de acumular hasta un 20% de su peso como hierro y que
presente en el citoplasma de estas
células, retenía más o menos del hierro dietario, dependiendo de las
necesidades de hierro del individuo. A este concepto se agregó a finales del
siglo XX, el descubrimiento del transportador de metales divalentes (DMT1) (72).
Este transportador, presente en la membrana apical de las células de la mucosa
intestinal, es el encargado de canalizar el hierro desde el lumen del intestino
al interior de las células de mucosa y cuya expresión en la membrana, aumenta o
disminuye de acuerdo con las necesidades de hierro. Desde un punto de vista celular,
estos conceptos, encontraron un apoyo muy importante en la función de las proteínas
dependientes de hierro (IRP1 y IRP2), ya que tal como se señaló anteriormente,
estas proteínas cuando hay hierro suficiente estimulan la síntesis de ferritina
y reducen la síntesis de DMT1, mientras que en deficiencia de hierro reducen la
síntesis de ferritina y estimulan la síntesis de DMT1 (23,66). Así,
por esta vía, las células de la mucosa podrían detectar el estado de hierro del
individuo y aumentarían la absorción de hierro en casos de deficiencia y la
disminuirían en casos de suficiencia.
El descubrimiento de la ferroportina y
su regulador negativo, la hormona hepcidina, durante el siglo XXI, enriquecen y
modifican notablemente estos conceptos y ofrecen un cuadro mucho más
convincente de cómo la mucosa intestinal regula la absorción del hierro. En este nuevo esquema, el sensor del estado
nutricional del hierro ya no es la mucosa intestinal sino el hígado (11),
que cuando detecta altos niveles de hierro circulante y celular produce y
secreta la hormona hepcidina (6,11,12,21).
Esta hepcidina viaja a través del suero y se une a su receptor celular la
ferroportina (18), estimulando su internalización y degradación. Como la ferroportina es el
único exportador celular del hierro (18), al disminuir su expresión
en la membrana celular, el hierro que ingresa a la célula no puede salir y se
queda atrapado en la célula depositándose en la ferritina que se sintetiza
libremente, ya que en condiciones de alto hierro disminuye la acción inhibitoria
ejercida por IRP1 o IRP2. Adicionalmente, en las células de la mucosa
intestinal el complejo hepcidina-ferroportina tiene un efecto negativo en la
expresión de DMT1 (18,22), con lo cual la captación del hierro
dietario también disminuye. El resultado neto de este proceso son células
epiteliales ricas en hierro que en lugar de ser liberado al suero, se pierden
en las heces, cuando las células del epitelio se descaman durante el proceso de
recambio celular, evitando una sobrecarga sistémica de hierro. En contraste, en
condiciones de bajo hierro el hígado no produce hepcidina por lo que la
captación del hierro dietario por DMT1 y la exportación del hierro captado por
la ferroportina aumentan para suplir las necesidades incrementadas de hierro o
compensar los efectos del consumo de una dieta baja en hierro absorbible. En
este caso, tal como se señaló anteriormente, la presencia de proteínas
sensibles al hierro (IRP1, IRP2) al estimular la síntesis de DMT1 y reducir la
síntesis de ferritina, contribuyen a amplificar el efecto de la baja hepcidina.
Otra observación con respecto al
metabolismo del hierro que no tenía una explicación convincente antes del
descubrimiento de la hormona hepcidina, era la disminución del hierro
circulante y la consecuente anemia del paciente infectado o con trauma e
inflamación. Esta condición es una ocurrencia permanente, de difícil
tratamiento y una señal de mal pronóstico en pacientes críticos (anemia del
paciente críticamente enfermo), que también se observa en pacientes con
enfermedades crónicas (anemia de la inflamación) y que es refractaria a los
tratamientos con hierro (46).
Los primeros reportes de esta situación
datan de finales de los años 20, pero el grupo de Cartwright y colaboradores en
el año 1946, fue uno de los primeros en describirla con rigurosidad y detalle (73).
Estos autores reportaron que en pacientes con endocarditis, pielonefritis,
tuberculosis, celulitis, septicemia, empiema, abscesos, osteomielitis y
neumonía, se producía una profunda hipoferremia y una anemia predominantemente
normocitica y normocrómica que no respondía al hierro ni al cobre, vitamina C,
cisteína, metionina o al consumo de hígado crudo. Al mismo tiempo reportaron
que después de inyecciones de ascorbato ferroso, en todos los pacientes
estudiados se detectaba un aumento en el hierro sérico que rápidamente disminuía
sin lograr corregir la anemia. Ya en esa época, estos investigadores postularon
que la anemia de la infección, resultaba de una incapacidad para sintetizar el
grupo hemo, debido a que no había hierro suficiente para incorporarlo en la
molécula de protoporfirina. Estos autores, atribuyeron la falta de hierro a una
alteración en el metabolismo intermediario del hierro que lo desviaba hacia los
depósitos, donde quedaba secuestrado e inaccesible al tejido eritropoyético. También
observaron que esta alteración era una característica de la infección, ya que
una vez corregida, los pacientes se recuperaban de la anemia. Estos mismos
autores, reprodujeron esta condición experimentalmente en perros utilizando el
producto de los drenajes de pacientes con osteomielitis crónica, infectados con
staphilococos aureus y pudieron reproducir en estos animales la hipoferremia y
anemia observada en los mismos pacientes. Además utilizaron muestras de
turpentina estéril, que produjo una respuesta inflamatoria que tuvo los mismos
efectos que ocasionó la infección sobre el metabolismo del hierro, lo que les
permitió concluir que era el tejido inflamado y no las bacterias, las
responsables de estos cambios (74). La etiología de esta condición
se desconocía, pero los autores ya en esa época proponían que el origen podía
generarse de algún producto derivado directamente del tejido inflamado, que era
también el sitio de una notable acumulación de hierro. Hoy se sabe que el
hierro se retiene principalmente en los macrófagos y en contraste con lo que
ocurre en organismos normales, no está disponible para la función
eritropoyética (46). También se acepta que esta condición puede
favorecer al paciente, ya que reduce el acceso al hierro a los microorganismos
invasores que lo necesitan para su sobrevivencia y reproducción (41).
Durante los últimos años del siglo XX,
hubo dos descubrimientos que aclararon en parte la hipoferremia de la
inflamación. El primero fue que la ferritina era una proteína de fase aguda que
secretan las células hepáticas en respuesta a citoquinas inflamatorias (IL-1, IL-6, FNT-α) (75) y un marcador de inflamación aguda y crónica que
aumenta en respuesta no específica en una serie de condiciones inflamatorias
como enfermedades crónicas del riñón, artritis reumatoidea y otras enfermedades
autoinmunes, infecciones agudas y neoplasias, con aumentos particularmente
notables en el caso de la enfermedad de Still y el síndrome hemofagocítico (76).
Con esto, los tejidos en respuesta a la inflamación, crean un espacio donde
depositar el hierro e incluso, la ferritina circulante, que aunque pobre en
hierro con respecto a la de depósito, puede contribuir a canalizar al hierro a
los depósitos, ya que los linfocitos, células hepáticas y otros tipos de
células tienen receptores para la ferritina (76), pudiendo así
contribuir a la hipoferremia de la inflamación.
La segunda revelación importante en
relación con la anemia de la inflamación fue el descubrimiento de la proteína
de resistencia natural asociada a los macrófagos o Nramp1 (77), que
es un transportador de hierro de la misma familia del Nramp2, conocido también
como DMT1 o DCT1 que es el transportador de hierro y otros cationes divalentes desde
el lumen del intestino a las células de la mucosa y un componente fundamental
en la captación del hierro en el ciclo de la transferrina en todas las células (23,66).
Nramp1 también está presente en todas las células, pero actúa sobre bacterias
invasoras intracelulares. Su función es extraer el hierro de estas células,
depletándolas de este metal esencial e impidiendo así su sobrevivencia y
multiplicación, confiriéndoles una capacidad de resistencia natural a las infecciones
(41,77). Además, en los macrófagos, se ha propuesto que la actividad
antimicrobiana de Nramp1 podría sustentarse en la capacidad del Fe(II) extraído
de los patógenos invasores para participar en las reacciones de Haber-Weiss o
Fenton y la inhibición del crecimiento de las bacterias por los radicales
hidroxilo generados en estas reacciones (78).
De la discusión anterior, no cabe duda
que tanto la ferritina como proteína de fase aguda, así como la capacidad de
Nramp1 de captar hierro, pueden contribuir a crear una situación de privación de
hierro para las bacterias infecciosas, generando una resistencia a la
propagación de la infección. Sin embargo, resulta más convincente que la
hipoferremia típica de la infección y de la anemia de la inflamación, la
produzca la hormona hepcidina, cuya síntesis es estimulada por citoquinas
inflamatorias, derivadas del proceso infeccioso e inflamatorio (Fig. 3). El
exceso de hepcidina al causar la endocitosis y degradación del único exportador
conocido del hierro celular, la ferroportina, atrapa al hierro en los
macrófagos, encargados de captar el hierro proveniente de los eritrocitos
senescentes y que movilizan unos 25 mg de hierro/día y en los enterocitos que
absorben entre 1-2 mg de hierro/día (23). Así, el atrapamiento
celular de este hierro (46), causa una disminución severa del hierro
circulante que produce anemia e impide que las bacterias invasoras dispongan
del hierro suficiente para su sobrevivencia y multiplicación y sirviendo además,
como un componente fundamental para la generación de radicales hidroxilos y la
destrucción de las bacterias en los macrófagos.
Conclusiones
Dentro del grupo de los micronutrientes
esenciales, el hierro tiene algunas particularidades que lo hacen único. La más
característica es que a pesar de ser uno de los metales más abundantes en la
corteza terrestre, es también el que causa las mayores deficiencias nutricionales. Así, la incidencia de deficiencia de
hierro y anemia, supera a las deficiencias de todos los demás micronutrientes(79,80,81).
Esto se debe a que el hierro en la naturaleza se encuentra principalmente en la
forma ferrica (Fe+++) como es el caso de los óxidos de hierro y el hierro
metálico, que son insolubles, mientras que el biológicamente activo, es el
hierro ferroso (Fe++), que es particularmente escaso y que el organismo protege
como un elemento traza, evitando su excreción. Adicionalmente, el hierro en
exceso es potencialmente tóxico y el umbral de toxicidad (~45mg/día) está
cercano a los requerimientos diarios de hierro en adultos (♀~18mg/día;
♂~8mg/día) (82), por lo que el organismo también limita su ingreso.
En relación con sus funciones, el hierro participa en el metabolismo de los
carbohidratos, proteínas, lípidos, así como en la síntesis y degradación de los
ácidos nucleicos, en la detoxificación de sustancias tóxicas, en las funciones
del sistema nervioso y del sistema inmune. Además, la deficiencia de hierro al
desactivar a la aconitasa provoca una acumulación de citrato que inhibe a la
fosfofructoquinasa-1 y a la vía glicolítica, direccionando así al metabolismo a
la utilización de grasa. Todo esto destaca su enorme importancia metabólica. Sin
embargo, la función más urgente del hierro está en la generación y utilización
de la energía necesaria para sobrevivir. En esto, la participación del hierro
se relaciona, tanto con el transporte del oxígeno asociado con la hemoglobina presente
en los glóbulos rojos, así como con la transferencia de los electrones provenientes
de la oxidación de los macronutrientes dietarios, a la molécula de oxigeno en
la vía de su reducción a agua, por medio del sistema de citocromos presentes en
la cadena respiratoria para producir ATP. Esta función del oxigeno y en
consecuencia del hierro que lo transporta y convierte en un generador de
energía, es tan decisiva que el ser humano puede sobrevivir semanas sin ingerir
alimentos, horas sin agua, pero sólo minutos sin oxígeno.
Esta dicotomía del hierro, entre su toxicidad y
esencialidad ineludible, requiere de un sistema homeostático riguroso y exacto que
logre mantener estas dos condiciones equilibradas. En este aspecto, los
descubrimientos de proteínas encargadas del transporte, y depósitos, así como
de las proteínas sensibles al hierro y sus efectos sobre transportadores y
receptores celulares del hierro durante el siglo XX, cimentaron las bases para
entender los aspectos regulatorios más importantes de la homeostasis de este
metal. Sin embargo, el descubrimiento de la hormona hepcidina y de su receptor la
ferroportina en los inicios del siglo XXI y sus efectos sobre la absorción y
redistribución del hierro, en condiciones de alto hierro, inflamación y anoxia
han representado un avance trascendental en el entendimiento de estos fenómenos.
Referencias
1.- Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary
antimicrobial peptide synthesized in the liver. J Biol Chem 2001; 276:
7806?7810.
2.- Hunter HN, Fulton DB,
Ganz T, Vogel HJ. The solution structure of human hepcidin, a peptide hormone
with antimicrobial activity that is involved in iron uptake and hereditary
hemochromatosis. J Biol Chem
2002; 277: 37597?37603.
3.- Ganz T. Hepcidin, a key regulator of iron
metabolism and mediator of anemia of inflammation. Blood 2003; 102: 783-788.
4.- Valore EV, Ganz T.
Posttranslational processing of hepcidin in human hepatocytes is mediated by
the prohormone convertase furin. Blood Cells Mol Dis 2008; 40 (1): 132-138.
5.- Pandur E, Nagy J, Po?r
VS, Sarnyai A, Husz?r A, Miseta A, Sipos K. "Alpha-1 antitrypsin binds preprohepcidin
intracellularly and prohepcidin in the serum".FEBS J 2009;276(7): 2012?2021.
7.- Kulaksiz H,
Theiligl F, Bachmann S, Gehrke S, Rost D, Janetzko A, Cetin Y, Stremme W. The
iron-regulatory peptide hormone hepcidin: expression and cellular localization
in the mammalian kidney. Journal of Endocrinology 2005; 184: 361?370.
9.- Urruita P, Aguirre P, Sparza A, Tapia U, Mena NP. Inflammation alters expression of DMT1,
FPN1 and hepcidin, and it causes iron accumulation in central nervous system
cells. J Neurochem 2013; 126: 541-549.
10.- Audin S, Celik O, Gurates B, Ulas M, Yilmaz M, Kalayci M,
Kuloglut T, Cataz Z, Ozercau JH, Kumru S. Concentrations of preptin, salusin
and hepcidin in plasma and milk of lactating women with and without gestational
diabetes mellitus. Peptides2013; 49: 123-130.
11.- Meynard D,
Babbitt JL, Lin HY. The liver: conductor of systemic iron balance. Blood 2014;
123:168-176.
12.- Fung E, Nemeth E. Manipulation of the hepcidin
pathway for therapeutic purposes. Haematologica 2013; 98: 1667-1676.
13.- Pigeon C, Ilyng G,
Courselanud B. A new mouse liver-specific gene encoding a protein homologous to
human antimicrobial peptide hepcidin, is overexpressed during iron overload. J
Biol Chem 2001; 276: 7811-8919.
14.- Nicolas G, Bennoun M,
Devaux I , Beaumont C, Grandchamp B, Kahn A, Vaulont S. Lack of hepcidin gene
expression and severe tissue iron overload in upstream stimulatory factor 2
(USF2) knockout mice. Proc Natl Acad Sci USA2001; 98 (15): 8780-8785.
15.- Nicolas G, Bennoun M,
Porteu A, Mativet S, Beaumont C, Grandchamp B, Sirito M, Sawadogo M, Kahn A,
Vaulont S. Severe iron deficiency
anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci USA 2002; 99 (7): 4596-4601.
16.- Nemeth E,
Ganz T. Regulation
of iron metabolism by hepcidin. Ann Rev Nutr
2006; 26: 323-342.
17.- Nemeth E, Tuttle MS,Powelson J, Vaughn M, Donovan
A,McVey Ward D. Hepcidin regulates cellular
iron eflux by binding to ferroportin (Fpn) and inducing its internalization.
Science 2004; 306: 2090-2093.
18.- Ward D, Kaplan J. Ferroportin-mediated iron
transport expression and regulation. Biochim Biophys Acta 2012; 1823(9):
1426-1433.
19,- Donovan A, Lima CA, Pinkus JL, Pinkus
GS, Zon LI, Robine S, Andrews NC: The iron exporter ferroportin/Slc40a1 is
essential for iron homeostasis. Cell Metab 2005; 1: 191?200.
20.- Fung E, Sugianto P, Hsu J, Damoiseaux R, Ganz T,
Nemeth E. High-throughput screening of small molecules identifies hepcidin
antagonists. Mol Pharmacol 2013; 83: 681-890.
21.- Zhao N, Zhang A, Enns C. Iron regulation
by hepcidina. J Clin Invest 2013; 123(6): 2337-2343.
22.- Chung B,
Chaston T, Marks J, Kaila S, Sharp PA. Hepcidin decreases iron transport
expression in vivo in duodenum and spleen and in vitro in THP-1 macrophages and
in intestinal Caco 2 cells. J Nutr 2009; 139: 1457-1462.
23.- Wessling-Resnick
M. Iron. En: Modern Nutrition in Health and Disease. Edición 11. Editores: Ross
C, Caballero B, Cousins RJ, Tucker KL, Ziegler TR. Lippincott Williams &
Wilkins. Baltimore MD USA 2012, pp 176-188.
24.- Nicolas G,
Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, Beaumont C, Kahn A, Vaulont
S. The gene encoding the iron regulatory peptide hepcidin is regulated by
anemia, hypoxia, and inflammation. J
Clin Invest 2002; 110(7):
1037-1044.
25.-
Delaby C, Pilard N, Goncalves AS, Beaumont C, Canonne-Hergaux F. Presence of
the iron exporter ferroportin at the plasma membrane of macrophages is enhanced
by iron loading and down-regulated by hepcidin. Blood 2005; 106: 3979-3984.
26.- Ganz T. Hepcidin and
its role in regulating systemic iron metabolism. Hematology 2006; 29-35.
27.- Ganz T. Hepcidin and iron regulation, 10 years later. Blood 2011; 117: 4425-4433.
28.- Sosa-Garrocho
M, Macías Silva M. El factor de crecimiento transformante Beta (TGF-?):
Funciones y vías de transducción. REB 2004;
23(I): 3-11.
29.- Goswani T, Andrews NC. Hereditary
hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests
a molecular mechanism for mammalian iron sensing. J Biol Chem 2006; 281(39): 28494-28498.
30.- Meynard D, Kautz
L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth MP. Lack of the bone
morphogenetic protein BMP6 induces iron overload. Nat Genet 2009; 41(4): 478-481.
31.- Andriopoulos B Jr,
Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, y col. BMP6 is a key
endogenous regulator of hepcidin expression and iron metabolism. Nat Genet
2009; 41(4): 482-487.
32.-Arndt S, Maegdefrau U, Dorn C, Schardt K, Hellerbrand
C, Bosserhoff K. Intestinal BMP6, a master gene for the regulation of iron
homeostasis. Meeting Report. Z Gastroenterol 2011; 49: A1.
33.- Seidah NG, Prat A.
The biology and therapeutic targeting of the proprotein convertasas. Nature
Drug Discovery 2012; 11: 367-383.
34.- Rhodes CJ.
Processing of the insulin molecule. En: Diabetes Mellitus: A Fundamental and
Clinical Text. Third Edition. Lippincot Williams & Wilkins. 2004, pp 29-51.
35.-Geoffrey N, Hendy H, Benett PJ, Gibbs BF, Lazure C.
Proparathyroid hormone is preferentially cleaved by the prohormone convertase
furin. Jour Biol Chem 1955; 270: 9517-9525.
36.-Pandur E, Sipos K, Grama L, Nagy J, Poor VS, Setalo
G, Miseta A, Fekete Z. Prohepcidine binds to the HAMP promoter and
autoregulates its own expression. Biochem J 2013; 451: 301-311.
37.-Peslova G, Petriak J, Kuzelova K, Hrdy I, Halada P, Kuchel PW, Soe-Lin Sh, Ponka P,
Sutack R, Becker E, Li-Hsuen H, Suryo Rahmanto Y, Richardson DR, Vyoral D.
Hepcidin the hormone of iron metabolism is bound specifically to α-2- macroglobulin in blood. Blood 2009;
113: 6225-6236.
38.- Fairbanks VF. Iron in medicine and nutrition. En: Modern
Nutrition in Health and Disease. Ninth Edition. Editores: Shils ME, Olson JA,
Shike M, Ross AC. Williams&Wilkins.
Philadelphia USA. 1999, pp 193-221.
39.-Wolf C.
Hiperpigmentación cutánea y homeostasis del hierro: Rol de la hepcidina. Rev Argent Dermatol 2007;
88: 96-104.
40.- McFarlane H, Reddy S, Adcock KJ, Adeshima A, Cooke R, Akene J.
Immunity, transferrin and survival in Kwasiorkor. Brit Med J 1970; 4: 268-273.
41.- Oppenheimer SJ. Iron and its relation to immunity and infectious
diseases. J Nutr 2001; 131: 616S-635S.
42.- Wang BH, Yu XJ, Wang D, Qi XM, Wang HP, Yang TT, Xu XH. Alterations
of trace elements (Zn,Se,Cu,Fe) and related metalloenzymes in rabbit blood
after severe trauma. J Trace Elem Med Biol 2007; 21 (2): 102-107.
43.- Weinberg ED. Infection and iron metabolism. Amer J Clin Nutr 1977;
9: 1485-1490.
44.- Besson-Fournier C, Latour Ch, Kautz L, Bertrand J, Ganz T, Roth
M-P, Coppin H. Induction of activin B inflammatory stimuli up-regulates
expression of the iron-regulatory peptide hepcidine through Smad 1/5/8
signaling. Blood 2012; 120: 431-439.
45.- Wrighting D, Andrewa N.
Interleukin-6 induces hepcidin expression through STAT3. Blood 2006;
108:3204-3209.
46.- Ganz T, Nemeth E. Iron
sequestration and anemia of inflammation. Semin Hematol
2009; 46(4): 387-393.
47.- Ruiz A, Brice?o O, Arteaga-Vizcaino M, Plumacher Z, Gonzélez M,
Quintero M. Anemia hemol?tica hereditaria y sobrecarga de hierro. VITAE.
Academia Biomédica digital 53: Enero Marzo 2013.
48.- Ke Q, Costa M. Hypoxia- induced factor
(HIF). Mol Pharmacol 2006; 70: 1469-1480.
49.- Harrison R. Structure and function of xanthine
oxidorreductase: Where are we now? Free
Radical Biol Med 2002; 33: 774-797.
50.-
Ramirez-Cheyne J, Zarante I. Deficiencia de glucosa-6-fosfato deshidrogenasa.
Situación actual, su relación con malaria y estrategias para calcular su
prevalencia. Univ Med Bogot?
2009; 50(1): 58-76.
51.- Gagliardo B, Kubat N, Faye A, Jaouen M, Deschemin JC, Canonne-Hergaux
F, Sari MA, Vaulont S. Pro-hepcidin is unable to degrade the iron exporter
ferroportin unless maturated by a furin-dependent process. J Hepatol 2009;
50(2): 394-401.
52.- Guillemot J, Canuel M, Essalmani R, Prat A, Seidah NG. Implication
of the proprotein convertases in iron homeostasis proprotein convertase 7 sheds
human transferrin receptor 1 and furin activates hepcidin. Hepatology. 2013;
57(6): 2514-2524.
53.- Hyuang-Keun K, Eun-Chul J, Ju-Ok Y,
Sun-Youg K, Hyunjuns- Ch, Sung-Soo K, Hiun-Suk Ch, Young-Seok Ch. Serum
prohepcidin levels are lower in patients with atrophic gastritis. Gastroenterol
Res and Pract 2013; 201810-201816.
54.- Ozcasap S, Yarali N, Isik P, Kava A,
Tune B. The role of prohepcidin in anemia due to Helicobacter pylori infection. Pediatr Hematol Oncol 2013; 30: 425-
431.
55.- Przbyszewska J, Zekanowska E,
Kedziora J, Kornatowka K, Boinska J, Pozynch K, Przybyszewski M, Cichon R.
Comparison of serum prohepcidin and iron metabolism parameters in obese and non
obese elderly individuals. Endokrynol Pol 2013; 64: 272-277.
56.- Kouiche H, Hirofumi U, Nobuchito O,
Tokinaga M, Utsonomiya A, Handa Sh, Hiroito T. Serum prohepcidin levels are
potential prognostic markers in patients with multiple mieloma. Experimental
Therapeutic Medicine 2012; 4: 581-588.
57.- Przbyszewska J, Zekanowska E,
Kedziora-Kornatowska K, Boinska J, Cichon R, Porzych K. Serum prohepcidin and
other iron metabolism parameters in anemia of chronic disease and with iron
deficiency anemia. Polskie Archivum Medycyny Wewnetrznej 2013; 123:105- 110.
58.-
Caliskan Y, Yelken B, Ozkok A, Gorgulu N, Yazici H, Telci A, Yildiz Z. Lower
serum prohepcidin levels associated with lower iron and erythropoyetic
requirements in hemodylaisis patients with chronic hepatitis C. BMC Nephrol 2012;
13: 56-63.
59.- Barrios Y, Espinoza M, Baron MA. Prohepcidina, su relación
con indicadores del metabolismo del hierro y de inflamación en pacientes
hemodializados tratados o no con eritropoyetina recombinante. Nutr Hospit 2010; 25: 555-560.
60.- Murphy AT, Witcher DR, Luan P, Wroblewski VS. Quantitation
of hepcidin from human and mouse serum using liquid chromatography tandem mass
spectrometry. Blood 2007; 110: 1048-1054.
61.-
Dudkowiak R, Neubauer K, Poniewierka E. Hepcidin and its role in inflamatory
bowel diseases. Adv Clin Exp Med 2013; 22: 585-591.
62.-
Oustamanolakis P, Koutrabakis IE, Kouroumalis EA. Diagnosing anemia in
inflamatory bowel disease: beyond the established markers. J Crohns Colitis
2010; 5: 381-391.
63.-
Oustamanolakis P, Koutroubakis IE, Messaritakis N, Malliaraki N, Sfiridakis A,
Kouromalis EA. Serum hepcidin and prohepcidin concentration in inflammatory
bowel disease. (8o Congress of European Crohnís and Colitis Organization Poster
P085. Viena Austria Feb 14-16, 2013.
64.-
Bunn HF. Eritropoietin. Cold Spring Harb Perspect Med 2013; 3: a011619.
65.- Drysdale JW, Munro HN. Regulation of
synthesis and turnover of ferritin in rat liver. Journ Biol Chem 1966;
241: 3630-3637.
66.- Wallander ML, Leibold EA, Eisenstein
RS. Molecular control of vertebrate iron homeostasis by iron regulatory
proteins Biochem Biophys Acta 2006; 1763: 668-689.
67.- Beinert H, Kennedy MC. Aconitase, a
two faced protein: enzyme and iron regulatory factor. FASEB
J 1993; 7: 1442-1449.
68.- Iron en Dietary Reference Intakes.
Institute of Medicine.Panel on Micronutrients. National Academy
Press. Washignton DC 2001.
69.- Frazer DM, Anderson GJ. I. Intestinal
iron absorption and its regulation. Am J Physiol, Gastrointestinal Liver
Physiol 2005; 289: G631-G635.
70.- Wood RJ, Ronnenberg AG. Iron. En: Modern Nutrition
in Health and Disease. Tenth Edition. Shils ME, Shike M, Ross AC, Caballero B,
Cousin RJ (Editores). Lippincott
Williams y Wilkins. Baltimore MD. 2006, pp 248-270.
71.- Laufberger V. Sur la cristallisation de la ferritine.
Bulletin de la Societe de chimie biologique 1937; 19: 1575-1582.
72.-
Cannone-Hergaux F, Gruenheid S, Ponka P, Gros P. Cellular and subcellular
localization of Nramp2 iron transporter in the intestinal brush border and
regulation by dietary iron. Blood 1999; 93: 4406-4417.
73.-
Cartwright GE, Lauritsen MA, Jones PJ, Merril IM, Wintrobe MM. The anemia of
infection. I Hypoferremia, hypercupremia and alterations in porphyrin
metabolism in patients. JCI 1946; 25: 65-68.
74.-
Cartwright GE, Lauritsen MA, Humphrey S, Jones PJ, Merrill IM, Wintrobe MM. The
anemia of infection. II The experimental production of the hipoferremia and
anemia in dogs. JCI 1946; 25: 81-86.
75.-
Tran TN, Eubanks SK, Schaffer KJ, Zhou CY, Linder MC. Secretion of ferritin by
rat hepatoma cells and its regulation by inflammatory cytokines. Blood 1997;
90: 4979-4986.
76.-
Wang W, Knovich MA, Coffman LG, Torti FM, Torti SV. Serum Ferritin: Past
present and future. Biochem Biophys Acta 2010; 1800: 760-769.
77.-
Barton CH, Biggs TE, Baker ST, Bowen H, Atkinson PG. Nramp1: a link between
intracelular iron transport and innate resistance to intracelular pathogens. Journal
of Leucocytes Biology 1999; 66: 757-762.
78.-
Zwilling BS, Kuhn DE, Wikoff L, Brown D, Lafuse W. Role of iron in
Nramp1-mediated inhibition of mycobacterial growth. Infect Immun 1999; 67:
1386-1382.
79.-
WHO. Worldwide prevalence of Vitamin A deficiencies in populations at risk
1995-2005. WHO global database on vitamin A deficiency. Geneva World Health
Organization 2006.
80.-
WHO, CDC. Worldwide prevalence of anemia 1993-2005. WHO Global database on
anemia. Geneva. World Health Organization 2006.
81.-
WHO Global database on Iodine deficiency. World Health Organization 2007.
82.-
Iron. En: Dietary Reference Intakes for: Vitamin A, Vitamin K, Arsenic, Boron,
Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon,
Vanadium and Zinc. Food and Nutrition Board. Institute of Medicine. National
Academy Press.Washington DC 2002.
NOTA:Toda la información que se brinda en este artículo es de cará
cter investigativo y con fines académicos y de actualización para estudiantes y profesionales de la salud. En ningún caso es de carácter general ni sustituye el asesoramiento de un médico. Ante cualquier duda que pueda tener sobre su estado de salud, consulte con su médico o especialista.