Dra. María Eugenia Orellana
Sección de Oftalmopatología Dr. Rafael Cordero Moreno y Dra. Malaquita de Cordero Moreno, Instituto Anatomopatológico Dr. José A. ODaly, Facultad de Medicina, Universidad Central de Venezuela
Lic. Carmen Abreu
Sección de Oftalmopatología Dr. Rafael Cordero Moreno y Dra. Malaquita de Cordero Moreno, Instituto Anatomopatológico Dr. José A. ODaly, Facultad de Medicina, Universidad Central de Venezuela
Dr. Rházes López
Servicio de Oftalmología, Hospital General de Este Dr. Domingo Luciani
Dr. Pablo Quintero
Servicio de Oftalmología, Hospital General de Este Dr. Domingo Luciani
Anatomía Patológica Retinoblastoma avanzado: hallazgos histopatológicos postratamiento Fecha de recepción: 03/04/2019
Fecha de aceptación:
10/06/2019
El retinoblastoma es el tumor intraocular más común de la infancia y el segundo tumor intraocular primario más frecuente en cualquier grupo etario. Objetivo: describir y comparar las características del retinoblastoma en casos que recibieron tratamiento y en no tratados. Métodos: estudio retrospectivo - descriptivo. Resultados: en los casos tratados, la mayoría tuvieron crecimiento mixto, abundante calcificación y siembras tumorales. A diferencia de los casos sin tratamiento previo en los cuales fue más frecuente el crecimiento endofítico, calcificaciones de ausentes a moderadas, y siembras tumorales de tipo polvo - esférulas - nubes. Con Chi-cuadrado no hubo diferencias significativas y con el test de U Mann Whitney para comparar los tipos de siembras, no hubo significancia estadística. Conclusión: los hallazgos fueron similares en ambos grupos, sin mostrar significancia estadística en las variables. Es de importancia el reporte de los hallazgos mencionados para la descripción, estadificación histopatológica y consecuente pronóstico de los pacientes.
Title Advanced retinoblastoma: post treatment histopathological findings
Abstract
Retinoblastoma is the most common intraocular tumor in childhood and the second common intraocular primary tumor in any age group. Objective: The aim was to describe and compare characteristics in retinoblastoma cases treated or not. Methods: descriptive retrospective study. Results: the majority of treated cases had mixed growth pattern, extensive calcification and tumor seedings. Non treated cases showed endophytic growth pattern, absence or moderate calcification and tumor seedings. Chi square were non-significant as well as U Mann Whitney test. Conclusion: histopathological findings were similar in both groups, without statistically significant data. Description of morphologic changes is important for histopathological classification, patient staging and prognosis.
Key Word retinoblastoma, pathology, oncology, treatment, tumor
Introducción
El retinoblastoma es el tumor intraocular más común de la infancia y el
segundo tumor intraocular primario más frecuente en cualquier grupo etario, esta
es una neoplasia de origen neuroectodérmico extraordinariamente radiosensible.
(1)
Este tumor embrionario de origen retinal, se presenta generalmente en
niños menores de 4 años, puede ser causa de ceguera e incluso muerte si no se
trata adecuadamente. Su incidencia es baja, de un caso por cada 15.000-20.000
nacidos vivos. Su diagnóstico, junto a su frecuente carácter hereditario (en un
40-50% de los casos se transmite con carácter autosómico dominante con alta
penetrancia), han hecho del retinoblastoma un paradigma del cáncer hereditario.
(2)
En globos oculares tratados con enucleación primaria, los hallazgos más
comunes son tumores neuroblásticos que forman pseudorosetas perivasculares,
compuestos por células de escaso citoplasma y núcleo hipercromático, que pueden
formar estructuras de diferenciación (rosetas de Homer Wright, rosetas de
Flexner Wintersteinery floretas), aun
cuando tienden a ser poco diferenciados, con patrón de crecimiento mixto, con
la posibilidad de invadir estructuras oculares (nervio óptico y coroides),
siendo este último elemento, un factor pronóstico de importancia. (3, 4)
Cuando se detecta tempranamente, se ubica entre los cánceres más
curables. El diagnóstico precoz y el reconocimiento de las posibles
implicaciones genéticas para el paciente y la familia son importantes para el
manejo y consejo genético óptimo. (5)
Los objetivos planteados en este estudio fueron describir las
características histopatológicas del retinoblastoma en aquellos casos que
recibieron algún tipo de tratamiento, así como comparar los hallazgos
histopatológicos en los casos de retinoblastoma tratados, con aquellos que no
recibieron terapia neoadyuvante.
Métodos
Se realizó un estudio retrospectivo descriptivo, donde se estudiaron
las características histopatológicas observadas en los casos de retinoblastoma
recibidos en el Instituto Anatomopatológico de la U.C.V., en cinco años
consecutivos que recibieron algún tipo de terapia neoadyuvante previa a la
enucleación. La investigación se realizó en la Sección de Oftalmopatología Dr.
Rafael Cordero Moreno y Dra. Malaquita de Cordero Moreno del Instituto
Anatomopatológico Dr. José A. O'Daly de la Facultad de Medicina de la Universidad
Central de Venezuela.
Para la realización de este trabajo, se seleccionaron y se recolectaron
los casos de retinoblastoma de los archivos de la Sección en un período de
cinco años, obteniéndose un total de 85 globos oculares de 82 pacientes. Se
reportaron los datos de los pacientes y la información demográfica se tabuló.
De las boletas de referencia se obtuvieron la edad al diagnóstico, género,
lateralidad, tiempo de evolución y tratamiento recibido, de ser el caso. Los
especímenes de enucleación fueron fijados en formalina buffer al 10%,
seccionados, procesados e incluidos en parafina para su posterior corte y
teñido para microscopía de luz. Se revisaron las láminas histológicas, coloreadas
con Hematoxilina Eosina, PAS (ácido peryódico de Schiff) y tricrómico de
Gomori.
Los parámetros histopatológicos estudiados fueron: patrón de
crecimiento, grado de diferenciación, necrosis, hemorragia, calcificación,
índice mitótico y apoptótico, basofilia de las paredes vasculares, siembras
tumorales, así como el grado de invasión de las capas oculares (de acuerdo a la
Clasificación Internacional).(6)(Tabla
1)
Los patrones de crecimiento incluyeron cinco subtipos: endofítico en el cual el tumor crece de
las capas internas de la retina hacia la cavidad vítrea. El patrón exofítico que crece principalmente de
las capas retinianas externas hacia el espacio subretiniano, ocasionando
desprendimiento de retina, pudiendo, en algunos casos, reemplazar el EPR
(epitelio pigmentado de la retina) e infiltrar la coroides a través de la ruptura
de la membrana de Bruch. El patrón mixto
constituye una mezcla de los dos patrones descritos previamente. El
retinoblastoma difuso donde el tumor
sustituye la retina en forma de un engrosamiento universal de la misma y simula
procesos inflamatorios; por último, el patrón
en regresión, en el cual el tumor se acompaña de una severa reacción
inflamatoria que conduce a ptisis bulbi. (7, 4)
El grado de diferenciación fue clasificado de acuerdo el porcentaje de
estructuras de diferenciación presentes (rosetas de Flexner
Wintersteinery floretas), siendo
considerado un tumor bien diferenciado cuando más del 80% del tumor presentaba
estas estructuras y poco diferenciado cuando estaban ausentes. El resto de los
tumores fueron clasificados como moderadamente diferenciados. (3)
La presencia de necrosis, hemorragia y calcificación se reportó como
ausente (0), poca (+), moderada (++) y extensa (+++). El índice mitótico y
apoptótico se obtuvo al sumar la cantidad de mitosis o cuerpos apoptóticos
observados en diez campos de mayor aumento, siendo clasificados en menos de 5,
entre 6 10 y más de 11 (en 10 campos de 400X).
Las siembras tumorales se reportaron en la cavidad vítrea, en el espacio
subretiniano, en la cámara anterior y en la cámara posterior; siendo descritas
como polvo, que provienen de
infiltración celular simple, las esférulas
que se originan de la expansión clonal de las siembras tipo polvo, estas pueden
ser traslúcidas clínicamente o pueden tener un centro blanquecino
correspondiente a necrosis rodeadas de células tumorales en mono o multicapas,
y las nubes que se forman a
consecuencia del desplazamiento del contenido del tumor primario. (8)
Resultados
En la Sección de Oftalmopatología del Instituto Anatomopatológico, en el
quinquenio seleccionado se recibieron 2218 especímenes, de los cuales 124
correspondieron a globos oculares, 85 de éstos tuvieron el diagnóstico
definitivo de retinoblastoma.
De los 85 globos oculares de 82 pacientes observados en dicho período,
la edad promedio fue de 27,6 meses, con un rango entre 3 120 meses. La
distribución por sexo fue de 32 del género femenino y 50 del masculino, con una
relación 1:1,5. El lado más frecuentemente afectado fue el izquierdo (54%). Se
conoció que 22 casos (27%) fueron bilaterales. El tiempo de evolución promedio
fue de 10,6 meses (rango 2 36 meses), entre los casos donde fue consignada
esta información. La aplicación de terapia neoadyuvante fue reportada en 29
casos y consistió de quimioterapia sistémica (tres drogas: carboplatino,
vincristina y etopósido) en 17 casos (59%), quimioterapia sistémica y
radioterapia en 5 casos (17%), quimioterapia sistémica y quimioterapia local en
2 casos (7%), quimioterapia sistémica + quimioterapia local + radioterapia en 2
casos (7%) y un caso de cada uno de los siguientes grupos de tratamiento:
quimioterapia sistémica + radioterapia + láser + crioterapia, quimioterapia
sistémica + crioterapia + radioterapia y crioterapia + láser.
Para el presente estudio, se seleccionó una cantidad similar de casos
que habían recibido tratamiento previo como de casos cuya enucleación fue
considerada como primaria, para un total de 12 globos oculares por grupo, cuyos
datos demográficos se reflejan en la tabla 1.
Tabla 1: Datos demográficos de casos de retinoblastoma
En los casos tratados, la mayoría de los mismos tuvieron patrón de
crecimiento mixto, escasa hemorragia, abundante calcificación, un número
elevado de cuerpos apoptóticos, escasas mitosis y siembras tumorales en la
mitad de los casos (Fig. 1 y 2).
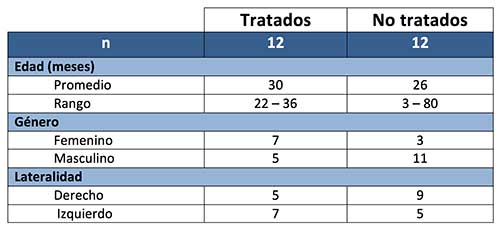
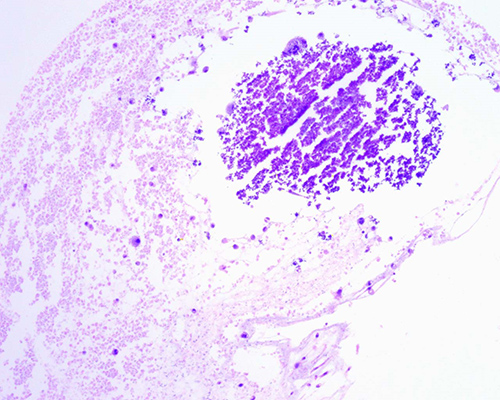
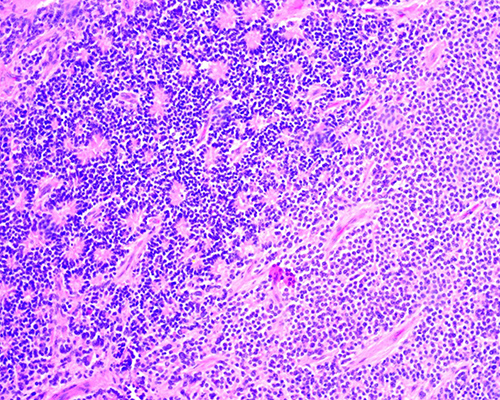
Fig.
1. Retinoblastoma tratado con ocho ciclos de
quimioterapia. Se aprecian abundantes calcificaciones distróficas (*)
intratumorales en el espesor de un tumor bien diferenciado. (H-E 40X)
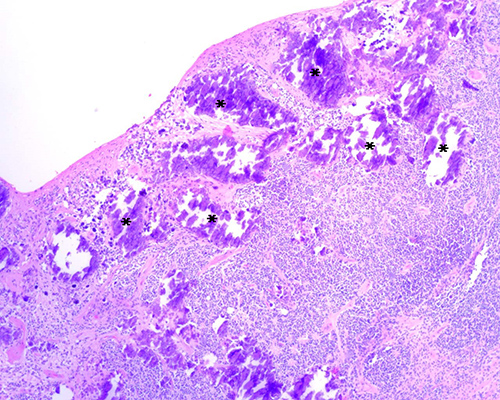
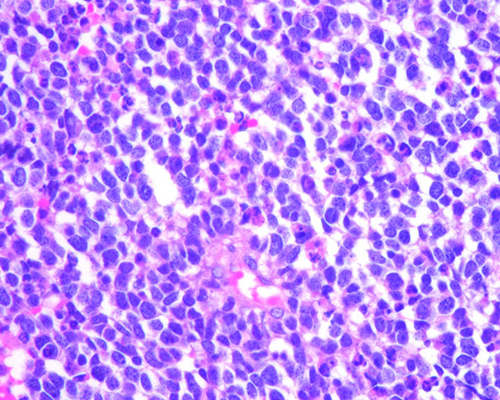
Fig.
2. Retinoblastoma tratado con tres ciclos de
quimioterapia. Se trata de un tumor poco diferenciado con abundantes cuerpos
apoptóticos (*). (H-E 400X)
A diferencia de los casos sin tratamiento
previo (enucleación primaria) en los cuales fue más frecuente el patrón de
crecimiento endofítico, hemorragia escasa a moderada, calcificaciones de
ausentes a moderadas en igual cantidad, y siembras tumorales de tipo polvo,
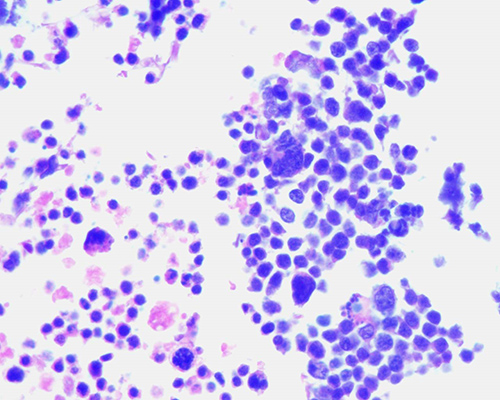
esférulas y nubes simultáneamente (Fig. 3 y 4).
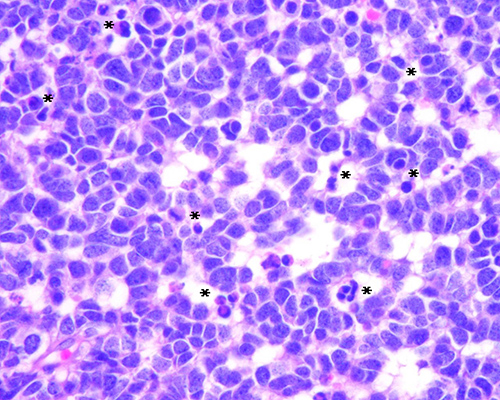
Fig.
3. Retinoblastoma no tratado. Se observa la
formación de pseudorosetas peritumorales rodeadas de extensa necrosis y
hemorragia. (H-E 100X)
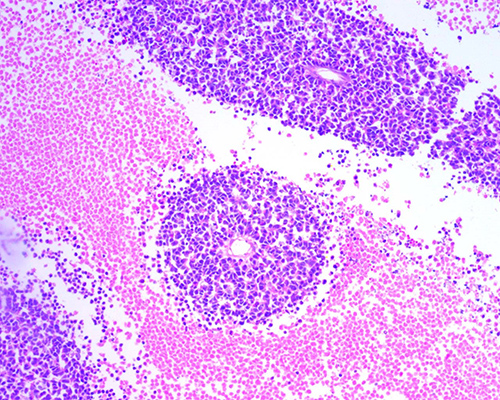
Fig. 4. Retinoblastoma tratado con cinco ciclos de quimioterapia. Presencia de siembras vítreas en forma de esférulas rodeadas de material necrótico. (H-E 100X)
De igual manera se observó el
patrón en regresión (ptisis bulbi) en dos casos para cada grupo. En ambos grupos se vieron en forma similar la
escasa diferenciación, necrosis moderada, abundantes cuerpos apoptóticos y
escasas mitosis (Fig. 5), así como la presencia de células viables en la mayoría de los casos tratados previamente (83% de los casos).
Fig.
5. Retinoblastoma bilateral tratado con tres ciclos
de quimioterapia. Se observa un tumor poco diferenciado con abundantes cuerpos
apoptóticos y escasas mitosis (H-E 400X)
Resaltamos la
presencia de reacción xantogranulomatosa en ambos grupos, caracterizada por la
presencia de abundantes histiocitos espumosos acompañados por escasas células
gigantes que se observaron tanto en el espesor del tumor como en estructuras
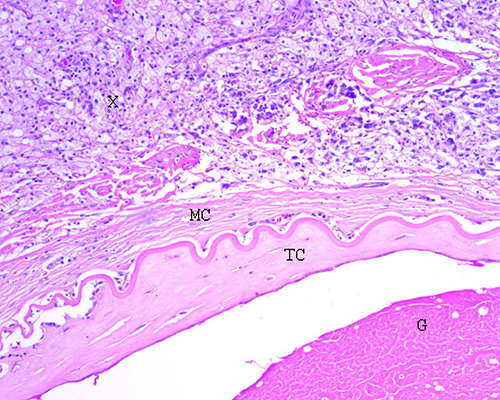
superficiales del globo ocular como la conjuntiva y epiesclera (Fig. 6).
Fig. 6. Retinoblastoma tratado con quimio y radioterapia. Se aprecia una reacción xantogranulomatosa (X) caracterizada por abundantes histiocitos espumosos ubicados en la cámara anterior. Presencia de membrana ciclítica (MC) que reviste la cara anterior del cristalino. Se reconoce la cápsula cristaliniana plegada, proliferación de tejido conectivo subyacente (TC) y glóbulos morganianos (G) en la corteza del cristalino constituyendo una catarata subcapsular anterior. (H-E 100X)
En los
dos grupos (un caso cada uno) se observaron células con atipia marcada y
aspecto bizarro (Fig. 7).
Fig.
7. Retinoblastoma tratado con quimioterapia
sistémica e intratecal. Se observan células con anisocitosis y anisonucleosis
marcadas, así como células gigantes tumorales de aspecto bizarro. (H-E 400X)
Igualmente se apreciaron zonas muy bien diferenciadas
caracterizadas por células de citoplasma moderado, núcleos redondos con
cromatina laxa, desprovistos de mitosis o apoptosis y con tendencia a la
formación de floretas que fueron descritas como áreas intratumorales de
retinocitoma, tanto en el grupo de retinoblastomas post tratamiento como en
los casos no tratados previamente (Fig. 8).
Fig. 8. Retinoblastoma tratado con ocho ciclos de quimioterapia. Se observan extensas áreas de tumor bien diferenciado (superior e izquierdo) con formación de rosetas de Flexner Wintersteiner, rosetas de Homer Wright y floretas, adyacente a un área de retinocitoma (inferior y derecho) caracterizado por células desprovistas de atipias, con citoplasma amplio eosinófilo y núcleo eucromático. (H-E 100X)
En cuanto al grado de infiltración
de las cubiertas del globo ocular, en los casos tratados fue predominante la
infiltración prelaminar del nervio óptico. En los casos no tratados, se vio
igual frecuencia de globos oculares sin infiltración del nervio óptico y con
presencia de tumor en la región retrolaminar con borde de resección negativo.
En ambos grupos se observó que la infiltración masiva de la coroides (o del
tracto uveal, en algunos casos) y la ausencia de tumor en la esclera fue lo más
observado (Fig. 9 y 10) (Tabla 2).
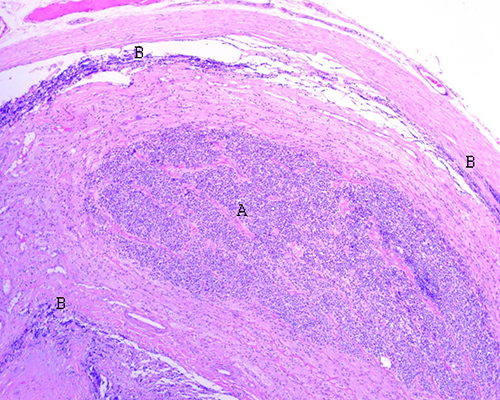
Fig.
9. Retinoblastoma tratado con cinco ciclos de
quimioterapia. Se aprecia la infiltración tumoral del nervio óptico en el
espesor del mismo (A) y en las cubiertas externas (B) (porción interna de la
duramadre, aracnoides y piamadre) (H-E 100X).
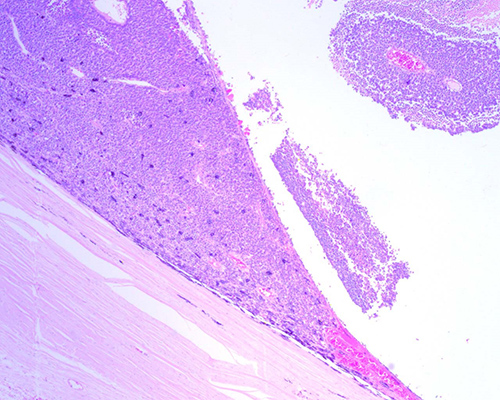
Fig. 10. Retinoblastoma no tratado. Se aprecia la infiltración masiva de la coroides y la indemnidad de la esclera. (H-E 40X).
Se aplicó la prueba de Chi-cuadrado para contrastar los datos observados
en ambos grupos (no tratados vs tratados), en las siguientes variables
(características macroscópicas y microscópicas): patrón de crecimiento, grado
de diferenciación, necrosis, hemorragia, calcificaciones, número de cuerpos
apoptóticos, número de mitosis y grado de infiltración de las cubiertas
oculares (según la Clasificación Internacional) (6), siendo los
resultados no estadísticamente significativos (p = 0,01) y en el caso de las
siembras tumorales, se aplicó el test de U Mann Whitney para comparar la
presencia de los tres tipos de siembras en ambos grupos, sin encontrar
significancia estadísticaya que el
valor de U fue 32, el valor crítico de U con p < 0,01 es de 9. (9)
Discusión
El retinoblastoma es un tumor maligno que se desarrolla a partir de la
retina y que por lo general aparece antes de los 5 años de edad. Es el tumor
intraocular más común en la infancia (1,5) y el segundo tumor
intraocular primario más común en cualquier grupo etario. (1) La
incidencia es más alta en países desarrollados y en algunos países de Centro y
Sur América, siendo uno de los tumores sólidos malignos más comunes en niños. (10)
Actualmente el enfoque terapéutico se basa en la quimiorreducción como
tratamiento inicial para la disminución del tamaño tumoral (11, 12) y
así lograr su control final con métodos locales conservadores (fotocoagulación,
crioterapia, termoterapia y braquiterapia) evitando de ser posible la
radioterapia externa y la enucleación. (11) Se han reportado
resultados exitosos utilizando la quimiorreducción en conjunto con el
tratamiento local para evitar la realización de enucleación o los efectos
adversos que conlleva la aplicación de radioterapia externa, al menos en el 50%
de los pacientes con retinoblastoma bilateral, según Antoneli et al 2006. (12)
La quimiorreducción, acompañada de terapias focales de consolidación ha
emergido como la terapia conservadora más utilizada en el manejo del
retinoblastoma. Este método es efectivo para tumores grandes y pequeños, uni o
multifocales, exo y endofíticos. Esta terapia ha reemplazado la radioterapia
externa como el tratamiento de elección en retinoblastomas bilaterales. (11,
12)
Los patrones de regresión fueron descritos inicialmente posteriores a
radioterapia e incluyen: el tipo 0 donde el tumor desaparece completamente sin
dejar cicatriz, el tipo 1 el tumor es sustituido por una masa calcificada, el
tipo 2 aparece como una masa no calcificada, el tipo 3 se caracteriza por una
masa parcialmente calcificada y el tipo 4 donde se observa una cicatriz plana,
atrófica. (13) Según lo
observado en nuestra casuística, el 83% de los casos tratados previamente,
mostraron células viables, similar a lo reportado por Bechrakis et al quienes indican la presencia de
tumor activo residual en cuatro de cinco casos enucleados por retinoblastoma
avanzado que habían recibido quimiorreducción. (14) Dithmar et al indican en dos casos estudiados,
la presencia de una masa gliótica en regresión en un caso y en el otro, un
tumor parcialmente necrótico con áreas adyacentes de tumor viable, (15)
lo cual es similar a lo observado en nuestros casos. Además, se apreció en tres
de nuestros casos tratados y en uno de los no tratados previamente, reacción
xantogranulomatosa que consistió en la presencia de abundantes histiocitos
espumosos acompañados de células epitelioides y células gigantes ocasionales infiltrando
en forma extensa o focal la cavidad ocular; Gombos et al reportaron un caso de colesterolosis intraocular posterior a
quimioreducción. (13, 14, 15, 16)
Otro hallazgo llamativo fue la
presencia de áreas de retinocitoma con o sin retinoblastoma adyacente en los
dos grupos del estudio, que ha sido observada en la serie de Demirci et al, quienes añaden que, de diez casos
estudiados, todos mostraron evidencia de regresión tumoral, ocho con regresión
completa y de éstos, seis ojos exhibieron un componente residual, inactivo,
bien diferenciado compatible con retinocitoma. Estos hallazgos indican que la
determinación de la viabilidad del retinoblastoma en casos previamente tratados
es complicada. Shields et al proponen
que los principales factores que afectan la regresión de un retinoblastoma son
el tamaño tumoral y la localización. (13, 17)
Algunos autores describen que los
globos oculares con retinoblastoma, tratados con neoadyuvancia pueden mostrar
cambios de retinopatía proliferativa con desprendimiento regmatogéno de la
retina y hemorragia vítrea, lo cual puede hacer difícil la diferenciación entre
una proliferación vítreo retiniana temprana, de una recurrencia tumoral. En
nuestros casos no se observaron estos cambios. (18)
En cuanto a la infiltración del
nervio óptico en esta serie, los casos tratados tuvieron invasión de la porción
prelaminar del nervio óptico e invasión del margen quirúrgico en un número
similar de casos, mientras que, en los casos con enucleación primaria, la
cantidad de globos oculares con nervio óptico libre y con invasión retrolaminar
y borde libre fueron los mismos. Los resultados no fueron estadísticamente
significativos. Igualmente para el grado de invasión de coroides y esclera, los
datos fueron similares. De Souza Filho et
al reportan que, en su casuística, los casos tratados con enucleación
primaria presentaban mayor afectación del nervio óptico y de las cubiertas oculares
cuando se compararon con ojos tratados previamente. (3)
Un estudio histopatológico de ojos tratados exclusivamente con
quimioterapia mostró que tumores con componente bien diferenciado evidenciaron
menos contracción por la quimioreducción. La mayoría de especímenes aquí
descritos, tanto tratados como no tratados, eran poco diferenciados. Sólo tres
en cada categoría mostraron una buena diferenciación y ninguno de éstos mostró
regresión con contracción del globo ocular tipo ptisis bulbi. (17)
En cuanto a las siembras tumorales, tanto vítreas como subretinianas,
Wilson et al ha señalado que la
dispersión celular en forma de siembras puede ser inducida por la
quimioterapia: cuando el tumor regresa durante los ciclos iniciales de la
quimioterapia, éste puede fragmentarse y liberar siembras hacia la cavidad
vítrea. La persistencia de estas siembras pueden también representar
penetración inadecuada de la quimioterapia a los sitios avasculares en la
cavidad vítrea y en el espacio subretiniano. (19) En los casos no
tratados de nuestra serie se pudieron observar los tres tipos de siembras
tumorales en forma individual o simultánea, mientras que en los tratados
previamente, hubo mayoría de ausencia de siembras aunque se vieron en forma
aislada, las siembras tipo polvo, tipo esférulas y las combinaciones polvo
esférulas y polvo esférulas nubes, sin denotar diferencia estadísticamente
significativa al comparar los grupos.
En conclusión, en la serie de casos estudiada se
observaron hallazgos similares en ambos grupos, con leve predominio de algunas
características, sin mostrar significancia estadística en ninguna de las
variables comparadas. Sin embargo, es de importancia la descripción de los
hallazgos mencionados para la correcta descripción, consecuente estadificación
histopatológica por parte de los patólogos y determinación del pronóstico de
los pacientes.
Referencias bibliográficas
Sang DN, Albert DM. Retinoblastoma: Clinical and Histopathologic
Features. Human Pathology. 1982, 13; (2):133-147.
Alonso J, Palacios
I, Gámez A, Camino I, Frayle H, Menéndez I, et al. Diagnóstico molecular del
retinoblastoma: epidemiología molecular y consejo genético. Med Clin (Barc).
2006, 126; (11):401-5.
de Souza Filho JP,
Martins MC, Torres VL, Toledo AB, Lowen MS, Pires LA, Erwenne CM. Achados
histopatológicos em retinoblastoma. Arq Bras Oftalmol.
2005;68(3):327-31
Orellana ME,
Fernandes BF, Arean C, Pifano I, Al-Kndari A, Burnier MN Jr. Clinical pathologic study of a cohort of
patients with retinoblastoma from a developing country. J Pediatr Ophthalmol
Strabismus.2009;46(5):294-9.
Wilson W. Retinoblastoma. Pediatr Rev 2007,28;(1):37-38.
Chantada G, Doz F,
Antoneli C, Grundy R, Clare F, Dunkel I, et al. A Proposal for an International
Retinoblastoma Staging System. Pediatric Blood Cancer 2005; 47: 801-805.
Font RL, Croxatto JO, Rao NA. Tumors of the Eye and Ocular Adnexa. AFIP
Atlas of Tumor Pathology. Fourth Series. Fascicle 5. ARP Press. Washington
2006. pp 85 103.
Munier FL. Classification and
management of seeds in retinoblastoma. Ellsworth Lecture Ghent August 24th
2013. Ophthalmic Genet 2014;35(4):193207
Chintagumpala M, Chevez-Barrios P, Paisse E, Plon S,Hurwitz R.
Retinoblastoma: Review of Current Management. The Oncologist 2007,12:1237
1246.www.theOncologist.com
Martín N, Coll MD, García J, Sánchez J, Triviño E, Guitart M, Gil J.
Retinoblastoma. Ann Oftalmol,2001;9(2): 74-92.
Antoneli CB, Ribeiro KC, Steinhorst F, Novaes PE,
Chojniak MM, Malogolowkin M.
Treatment of retinoblastoma patients with chemoreduction plus local therapy:
experience of the AC Camargo Hospital, Brazil. J Pediatr Hematol Oncol 2006; 28
(6): 342-5
Shields CL, Palamar M,
Sharma P, Ramasubramanian A, Leahey A, Meadows AT, Shields JA. Retinoblastoma Regression Patterns Following
Chemoreduction and Adjuvant Therapy in 557 Tumors. Arch Ophthalmol. 2009;127(3):282-290
Bechrakis NE, Bornfeld N, Schueler A, Coupland SE,
Henze G, Foerster MH. Clinicopathologic features of retinoblastoma after
primary chemoreduction. Arch Ophthalmol. 1998;116(7):887-893.
Dithmar S, Aaberg
TM, Grossniklaus HE. Histopathologic changes in retinoblastoma after
chemoreduction. Retina. 2000;20(1):33-36.
Gombos DS, Howes E,
OBrien JM. Cholesterosis following chemoreduction for advanced
retinoblastoma. Arch
Ophthalmol 2000; 118 (3): 440-1.
Wilson MW,
Rodriguez-Galindo C, Haik BG, Moshfeghi DM, Merchant TE, Pratt CB. Multiagent
chemotherapy as neoadjuvant treatment for multifocal intraocular
retinoblastoma. Ophthalmology 2001; 108 (11):2106-14
NOTA:Toda la información que se brinda en este artículo es de cará
cter investigativo y con fines académicos y de actualización para estudiantes y profesionales de la salud. En ningún caso es de carácter general ni sustituye el asesoramiento de un médico. Ante cualquier duda que pueda tener sobre su estado de salud, consulte con su médico o especialista.