Freddy González-Mujica
freddygonzalezmujica@gmail.com
Médico-Cirujano, PhD en Bioquímica
Sección de Bioquímica Médica, Instituto de Medicina Experimental, Facultad de Medicina, Universidad Central de Venezuela.
Monografías docentes Resistencia a la insulina Fecha de recepción: 31/12/2000
Fecha de aceptación:
31/12/2000
La insulina es una hormona polipeptídica cuyo mecanismo de acción se inicia por su unión a un receptor de membrana (IR), a partir de lo cual se generan dos vías de señalización, una metabólica y otra mitogénica; la translocación a la membrana plasmática del transportador de glucosa 4 (GLUT4) forma parte de la vía metabólica, pero por su importancia, es considerada de manera independiente. La resistencia a la insulina es el resultado de que las células que normalmente responden a la hormona dejan de hacerlo adecuadamente. Alteraciones en las vías de señalización de la insulina condicionan resistencia a la hormona, dichas modificaciones pueden deberse a mutaciones y/o modificaciones post traduccionales de IR y/o de los efectores moleculares de las citadas vías. Se ha postulado que procesos tales como la inflamación, el estrés del retículo endoplasmático (ER), la disfunción mitocondrial y el hiperinsulinismo producen resistencia a la insulina. La obesidad se acompaña de inflamación crónica con la consecuente producción de citoquinas las cuales activan proteína quinasas de serina/treonina que fosforilan e inactivan a IR y a los substratos de IR (IRS) causando resistencia a la insulina. Durante el estrés del ER también se activan proteína quinasas de serina/treonina que inactivan IR e IRS. En la obesidad al igual que en la disfunción mitocondrial, se acumulan lípidos los cuales dan origen a caramidas y diacilgliceridos, los primeros activan proteína fosfatasas que defosforilan e inactivan a Akt/PKB, inhibiendo la vía de señalización de la insulina y los segundos activan proteína quinasas de serina/treonina que inactivan IR e IRS. Durante la disfunción mitocondrial se producen especies reactivas de oxígeno (ROS) las cuales activan proteína quinasas de serina/treonina, con las consecuencias antes mencionadas. Durante el hiperinsulinismo se activa un asa de retroalimentación negativa, por la cual las proteína quinasas de las vías de señalización de la insulina fosforilan en serina/treonina a IR e IRS ocasionando resistencia a la insulina.
Palabras Claves:Insulina, resistencia a la insulina, estrés del retículo endoplasmático, disfunción mitocondrial, hiperinsulinismo
Abstract
Insulin is a polypeptide hormone, whose mechanism of action begins by its binding to a membrane receptor (IR), from it starts two ways of signalling, one metabolic and the another mitogenic; the translocation to the plasma membrane of the glucose transporter 4 (GLUT4) is part of the metabolic pathway, but due to its importance is consider separately. The insulin resistance occurs when the cells that normally respond to the hormone stop doing so. Alterations in the insulin signal pathway produce hormone resistance; such modifications may be due to mutations and/or post-translations modifications of IR and/or of the molecules effectors of the above mentioned pathway. It has been postulated that the insulin resistance is the consequence of inflammation, endoplasmic reticulum (ER) stress, mitochondria dysfunction and/or hyperinsulinism. Obesity is accompanied of a low grade chronic inflammation with the consequence cytokines production, which activate serine/threonine protein kinase that phosphorylate and inactivate IR and its substrate (IRS) conditioning insulin resistance. Also, during the ER stress serine/threonine protein kinase are activated which in turn phosphorylate and inactivate IR and IRS. In the course of the obesity and in the mitochondrial dysfunction there is lipid accumulation which are used to produce ceramides and diacylglycerols, the first one activate protein phosphatase that dephosphorylate and inactivate Akt/PKB inhibiting the insulin signal pathway, and the second one activate serine/threonine protein kinase that inactivate IR and IRS. The mitochondrial dysfunction conditioning the production of reactive oxygen species (ROS) which activate serine/threonine protein kinase with the consequence above mentioned. The hyperinsulinism activates a negative feedback loop; in consequence the protein kinases of the insulin signal pathway phosphorylate in serine/threonine and inactivate IR and IRS producing insulin resistance.
Para la mayoría de los habitantes de este mundo, la disponibilidad de alimentos es escasa e impredecible, necesitándose el desarrollo de mecanismos eficientes para el adecuado uso y depósito de la energía. Sin embargo en nuestro mundo moderno, con una sociedad mecanizada, la demanda calórica se ha minimizado y por otro lado los alimentos y bebidas muy agradables y densos en calorías están fácilmente disponibles. Estas condiciones han permitido la aparición de la actual pandemia de obesidad y las condiciones asociadas: la enfermedad del hígado graso no alcohólico (NAFLD por sus siglas en inglés), la arterioesclerosis y la diabetes mellitus tipo 2 (DM2). La resistencia a la insulina es una característica común de estas enfermedades y una importante cantidad de esfuerzo se ha invertido tratando de delinear la patogenia de la misma. La resistencia a la insulina es el resultado de que las células que normalmente responden a la hormona dejan de hacerlo adecuadamente (1).
La DM2 es la enfermedad endocrina más frecuente del hombre y de acuerdo a la Federación Internacional de Diabetes (IDF por sus siglas en inglés), en el 2017 afectó aproximadamente a 425 millones de personas adultas alrededor del mundo (2). Evidencias experimentales y clínicas demuestran que la resistencia a la insulina de tejidos tales como el hepático, el adiposo y el muscular son la principal característica de ésta disfunción metabólica. La resistencia periférica a la insulina condiciona que las células β pancreáticas incrementen su producción condicionando hiperinsulinismo compensatorio lo cual agrava la resistencia a la hormona; la sobre estimulación de las células β pancreáticas por tiempo prolongado, aunado a la resistencia a la insulina conlleva a un agotamiento de las misma con la consecuente disminución de la producción de insulina, hiperglicemia sostenida y DM2 (3, 4). A nivel molecular, la resistencia a la insulina es la consecuencia de alteraciones en la vía de señalización de la hormona, debido a mutaciones o a modificaciones post-traduccionales del receptor de la hormona o de los efectores proteicos corriente abajo (5).
En general, varios mecanismos celulares intrínsecos y extrínsecos han sido reconocidos, los cuales muestran una relación causa efecto entre el aumento de peso corporal y la resistencia periférica a la insulina (6). Los mecanismos celulares intrínsecos incluyen: la disfunción mitocondrial, el stress oxidativo y el stress del retículo endoplasmático, mientras que las alteraciones en los niveles de adipoquinas y de ácidos grasos y la presencia de inflamación en los tejidos metabólicamente activos, son los mecanismos extrínsecos dominantes en la modulación de la acción de la insulina (6).
En el presente trabajo nos proponemos revisar los mecanismos moleculares propuestos hasta el presente para explicar la resistencia a la insulina, sin embargo creemos conveniente discutir primero las vías de señalización de la hormona y su regulación para poder comprender mejor como las alteraciones de las mismas condicionan resistencia a la insulina.
Mecanismo de acción de la insulina
La insulina es una hormona polipeptídica constituida por dos cadenas denominadas A y B de 21 y 30 aminoácidos respectivamente, producida y secretada por las células β de los islotes pancreáticos, la cual ejerce un efecto pleotrópico (7).
El mecanismo de acción de la insulina ha sido revisado recientemente (8) y aquí solo discutiremos los aspectos más relevantes. La acción de la hormona es mediada por un receptor de membrana (IR por siglas en inglés) el cual es un tetrámero formado por 2 subunidades α y 2 subunidades β con una estructura modular,un dominio extracelular formado por la totalidad de las subunidades α, las cuales están abundantemente glicosiladas y con un peso molecular de 135 KDa, y una pequeña proporción de la subunidades β, esta última se continua con un segmento incluido en la membrana plasmática seguido luego por el dominio citoplasmático, el peso molecular de la subunidad β es de 95 KDa. Existen puentes disulfuro entre las subunidades α y entre α y la porción extracelular de la subunidad β. En el dominio extracelular se encuentran los sitios de unión a la insulina y en el dominio citoplasmático radica la actividad de tirosina quinasa (9). Cuando IR se une a la insulina se promueve la autofosforilación del receptor y la fosforilación de substratos específicos de IR (IRS por sus siglas en inglés), de los cuales 2 son los más importantes IRS1 e IRS2, iniciándose fundamentalmente 2 redes de señalización, a saber: la vía del fosfatidilinositol-3-quinas (PI3K por sus siglas en inglés)/Akt también conocida como proteinquinasa B (PKB por sus siglas en inglés), la cual es responsable de la mayoría de los efectos metabólicos de la insulina y está conectado exclusivamente a IRS y la vía de Raf/Ras/MEK/MAPK (proteína quinasa activado por mitógeno también conocida como quinasa regulada por señales extracelulares, ERK por sus siglas en inglés) la cualparte de ambos IRS y proteínas Shc (proteínas adaptadoras con dominios de homología Src 2) y está relacionada con la regulación de la expresión genética y con la cooperación de PI3K en el control del crecimiento (mitogéneis) y diferenciación celular.
Vía de la Fosfatidilinositol-3-quinasa
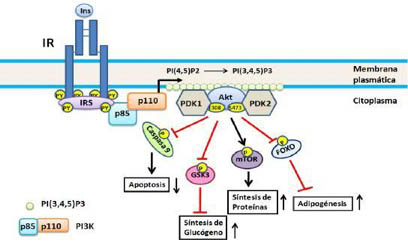
Esta vía (10) se inicia por la interacción de IRS1 y/o IRS2, fosforilados por IR, con PI3K activándose esta última, y en consecuencia produce fosfatidilinositol 3; 4; 5 trifosfato (PIP3) el cual funciona como un segundo mensajero activando a una proteína quinasa dependiente de fosfatidilinositol (PDK por sus siglas en inglés), ésta última fosforila y activa a Akt/PKB (Figura 1).
Figura 1 Vía del PI3K. Se inicia por la unión de PI3K a los IRS fosforilados por IR, activándose y en consecuencia produce PIP3, este último actuando como segundo mensajero activa a PDK la cual fosforila a Akt/PBK. En el esquema se representan cuatro de los substratos de Akt/PBK: caspasa 9, GSK3, mTOR y Fox0; no se incluye AS160 ya que ésta será objeto de consideración aparte. Para detalles ver el texto.
Akt/PKB es una familia de proteína quinasas de serina/treonina las cuales fosforilan una serie de proteínas entre las que destacan:
El blanco de la rapamicina en los mamíferos (mTOR por sus siglas en inglés) el cual al estar fosforilado, a su vez fosforila el factor de iniciación eucariota E4 y de la proteína ribosomal p70 S6 quinasa, estimulando la síntesis de proteínas.
La quinasa 3 de la glucógeno sintasa (GSK3 por sus siglas en inglés) que al estar fosforilada es inactiva y no puede fosforilar e inactivar a la glucógeno sintasa, estimulando la síntesis de glucógeno.
La familia 0 de los factores de transcripción cabeza de tenedor (Fox0 por sus siglas en inglés) y en particular Fox01 el cual al estar fosforilado no puede entrar al núcleo para activar los genes neoglucogénicos y adipogénicos.
Fosforila y activa a la enzima sintasa de óxido nítrico endotelial (eNOS por sus siglas en inglés) la cual produce la molécula vasodilatadora y anti-inflamatoria óxido nítrico (NO) estableciéndose una conexión potencial entre la resistencia a la insulina y las enfermedades cardiovasculares.
Fosforila la proteínas caspasa 9, inhibiendo su actividad apoptótica у promoviendo por tanto la supervivencia celular.
Translocación del GLUT 4 regulado por la insulina.
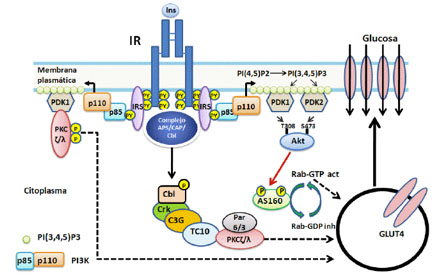
El efecto clásico de la insulina es el promover la entrada de glucosa a los tejidos adiposo, músculo esquelético y cardiaco, esto ocurre gracias a la translocación, desde vesículas intracelulares (GSV por sus siglas en inglés) a la membrana plasmática, del transportador de glucosa 4 (GLUT4) por un mecanismo que aún no está completamente claro (Figura 2).
Figura 2 Exocitosis de GLUT4. La insulina promueve la translocación de GLUT4 desde las GSV a la membrana plasmática. La proteína AS160 en su estado no fosforilado y activo estimula la actividad de GTPasa de la proteína G de bajo peso molecular Rab generando Rab-GDP el cual es inactivo, inhibiendo el tráfico de vesículas. Cuando AS160 es fosforilado por Akt/PBK se inhibe, incrementándose Rab-GTP y se incrementa el tráfico de vesículas. Por otra parte PDK fosforila la aPKC (ζ/λ) la cual incrementa la translocación de GSV. Se ha postulado una vía alterna que parte de IR el cual activaría a las proteínas APS-CAP-Cbl y estas activarían a la proteína G de bajo peso molecular TC10 que activaría aPKC (ζ/λ) la cual incrementa la translocación de GSV.
La principal vía involucrada en la translocación de las GSVs es la del PI3K PDK Akt/PBK, (Figura 2) ésta última fosforila uno de sus substrato AS160 la cual es una proteína que modula la actividad de GTPasa de la proteína de bajo peso molecular Rab. Cuando AS160 no está fosforilado es activa, estimula la actividad de GTPasa de Rab generándose Rab-GDP el cual es inactivo y se bloquea la exocitosis de GLUT4. Por el contrario la fosforilación de AS160, por Akt/PBK, la inactiva con lo cual predomina Rab-GTP incrementándose el tráfico de vesículas que contienen GLUT4 a la membrana plasmática (11).
Por otro lado, la proteína quinasa C atípica (aPKC por sus siglas en inglés) requiere de PIP3 y fosforilación por PDK pero no está relacionada Akt/PKB; está involucrada en la translocación de GLUT4 a la membrana plasmática y la captación de glucosa por los tejidos adiposo y muscular (Figura 2).
Se ha descrito una vía independiente de PI3K, que involucra CAP (proteína activada por catabolíto la cual une AMPc) y APS (proteína adaptadora que es substrato del IR) que interactúa con Cbl (proteína que une ubiquitina, está relacionada con la señalización de ubiquitinización y degradación de proteínas y posee un dominio que une tirosinas fosforiladas) en la translocación de GLUT4 y la captación de glucosa estimulada por la insulina (Figura 2). Clb es fosforilado en tirosinas por IR y en consecuencia recluta APS y CAP; posteriormente se une al complejo CrkII (proteínas adaptadoras que unen varios tipos de proteínas fosforiladas en tirosina además de poseer dominios SH2), esta a su vez interactúa con un factor liberador de nucleótidos de guanina (C3G) el cual se comporta como un factor intercambiador de la proteína de bajo peso molecular que une GTP, TC10 que activaría la aPKC (ζ/λ) con el consecuente incremento de la translocación de GSV a la membrana plasmática (Figura 2).
Vía de Raf/Ras/MEK/ MAPK
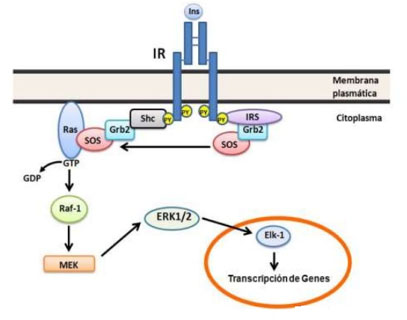
Una segunda rama esencial de la señalización de la insulina la constituye la vía de Raf/Ras/MEK/ MAPK (12), la cual es independiente de PI3K y de AKT/PKB (Figura 3). Ambos, IR activado y las proteínas IRS activadas poseen sitios de unión a proteínas que contienen dominios SH2 tales como Grb2 (proteína adaptadora que une al factor de crecimiento 2) y Shc (que poseen dominios de unión a fosfotirosina, PBT, y SH2). Grb2 une proteínas como Gab-1 (es uno de los substratos de las proteínas IRS) y proteínas ricas en prolina tales como hijo de los sin siete (SOS por sus siglas en inglés) la cual funciona como un intercambiador de nucleótidos de guanina para Ras (proteínas de bajo peso molecular que unen nucleótidos de guanina, controlan diferentes fenómenos: integridad del citoesqueleto; proliferación, diferenciación, adhesión y migración celular y la apoptosis). SOS permiten la conversión de Ras asociado a la membrana plasmática de la forma inactiva unido a GDP a la forma activa unido a GTP. Ras-GTP activa a la proteína quinasa de serina/treonina denominada Raf, la cual estimula por fosforilación, en serina/treonina, su blanco corriente abajo son MEK1 y 2 (este acrónimo deriva de MAP quinasa, ERK y Kinasa), estos a su vez fosforilan (en serina/treonina) y activan a las MAP quinasas (proteína quinasa activada por mitógeno) la cual activa por fosforilación a ERK1 y 2 (quinasa regulada por señal extracelular). La activación de ERK1 y 2 juega un papel directo en la proliferación y diferenciación celular regulando la expresión de genes y eventos extranucleares tales como la organización del citoesqueleto mediante la fosforilación y activación de blancos en el núcleo y el citoplasma.
Figura 3 Vía de Raf/Ras/MEK/ MAPK. Esta vía se inicia con la interacción de IR e IRS activados con las proteínas adaptadoras Grb2 y Shc, ésta última une SOS que actúa como un intercambiador de nucleótidos de guanina en la proteína de bajo peso molecular Ras. Ras-GTP fosforila y activa a la proteína quinasa Raf, ésta a vez fosforila a MEK, la cual activa por fosforilación a ERK 1 y 2 que modifican eventos nucleares y citoplasmáticos. Para detalles ver el texto.
Finalización de la señal y mecanismos de retroalimentación negativos
La activación de proteína quinasas por la insulina está sujeta a la acción opuesta de proteína fosfatasas y de mecanismos de retroalimentación negativos, el balance adecuado entre ambos procesos garantiza una apropiada sensibilidad a la insulina y su alteración condiciona resistencia a la acción de la hormona (8). A continuación pasaremos revista a los mecanismos más importantes de control negativo:
La fosforilación en algunas serinas específicas, de IRS condicionan que los mismos no sean reconocidos por IR y/o que no puedan unirse a PI3K o aún más que se promueva su degradación.
La actividad de proteína fosfatasas que actúen sobre tirosinas fosforiladas como la 1B (PTP1B por sus siglas en inglés) atenúan la respuesta a la insulina por promover la defosforilación de IR y de los IRS. Otro punto de control negativo lo constituye la defosforilación e inactivación de Akt/PBK y aPKC por proteína fosfatasas de serina/treonina 2A (PP2A por sus siglas en inglés).
La fosfatidilinositol-3; 5; 4; trisfosfato 3-fosfatasa (PTEN por sus siglas en inglés) y la fosfatasa de fosfoinositol -3; 5; 4; trisfosfato que contiene dominio SH2 (SHIP2 por sus siglas en inglés) son enzimas que hidrolizan PIP3 con lo cual se reducen la actividad de PDK y en consecuencia se inactivan Akt/PKB y aPKC disminuyendo los efectos de la insulina (Figuras 1 y 2).
Los IR, unido a la insulina, son internalizados por endocitosis mediada por clatrina para ser reciclados o degradados en los lisosomas.
Mecanismo molecular de la resistencia a la insulina
La característica más relevante de la DM2 es la resistencia a la insulina, una condición en la cual, las células que normalmente responden a la hormona, dejan de hacerlo adecuadamente (13). Esta deficiencia en la respuesta a la insulina es causada por diferentes alteraciones, incluyendo mutaciones y/o modificaciones postraduccionales de IR, IRS o de los efectores moleculares de la cadena de señalización de la hormona. Las alteraciones más comunes que condicionan resistencia a la insulina son: disminución en la cantidad y/o en la actividad catalítica de IR, un aumento en el estado de fosforilación en serina/treonina de IR y/o IRS, aumento de la actividad enzimática de fosfatasas de tirosinas fosforiladas, fundamentalmente PTP 1B, las cuales actúan defosforilando e inactivando IR e IRS, disminución de la actividad de quinasa de PI3K y Akt/PKB y defectos en la expresión y/o función del GLUT4 (14). Las alteraciones antes descritas condicionan que los tejidos adiposo y muscular capten menos glucosa y se produzca disfunción metabólica en los mismos.
Un aspecto fundamental de la resistencia a la insulina lo constituye la fosforilación de los IRS en restos de serina y treonina (15) con lo cual se disminuye su fosforilación en tirosina y en consecuencia se reduce su interacción con PI3K, esto a su vez condiciona que se reduzca la fosforilación y consecuente activación de Akt/PKB. Además se ha reportado que la fosforilación de los IRS en restos de serina y treonina aumenta la velocidad de su degradación. Varios agentes tales como: citoquinas pro-inflamatorias, ácidos grasos saturados, algunos aminoácidos, endotelina 1, angiotensina II y el hiperinsulinismo (16-18) estimulan la actividad de varias quinasas, tales como: varias isoformas de PKC, quinasa N terminal (JNK por sus siglas en inglés), mTOR, proteína quinasa ribosomal de 70 KDa S6 (p70 KDa S6K por sus siglas en inglés), proteína quinasa A (PKA por sus siglas en inglés) y MAPK las cuales fosforilan en restos de serina y treonina a los IRS (19). Las alteraciones antes descritas son suficientes para condicionar intolerancia a la glucosa, la cual puede progresar hasta DM2, especialmente si las células β pancreáticas son incapaces de producir una hiperinsulinemia compensatoria (20).
Por otro lado, el incremento en el metabolismo de ácidos grasos saturados condiciona un aumento en la producción de ceramidas (ácido graso unido a esfingosina), las cuales estimulan la actividad de la PP2A y ésta última desfosforila varias proteínas, entre las mismas destaca Akt/PKB (21), la cual se inactiva interfiriendo en una de las vías de señalización de la insulina (Figura 1 y 2).
Inflamación y resistencia a la insulina
La inflamación es un proceso fisiológico caracterizado por un aumento en el número de glóbulos blancos sanguíneos y/o citoquinas pro-inflamatorias en la circulación y/o en los tejidos (22). La obesidad se define como la excesiva cantidad de grasa corporal, la cual se manifiesta como un incremento en el peso corporal asociada a una más alta distribución del tejido adiposo hacia las vísceras (14). En la obesidad existe un bajo grado de inflamación crónica la cual está involucrada en la patogénesis de una serie de enfermedades, entre las cuales destacan: la DM2, la arterioesclerosis, el hígado graso no alcohólico y la hipertensión arterial. La inflación presente en el tejido adiposo repercute en los tejidos hepático y muscular lo cual condiciona resistencia a la insulina y disfunción metabólica sistémica. En los pacientes con obesidad se ha detectado un incremento en la cantidad de un alto número de marcadores de inflación y una estrecha correlación entre dichos marcadores y la adiposidad abdominal (23).
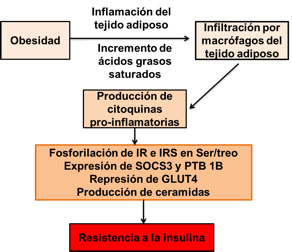
El incremento del tamaño de las células adiposas y la expansión del tejido adiposo, presente en la obesidad, condicionan alteraciones en la secreción de adipoquinas (citoquinas producidas por el tejido adiposo), citoquinas pro-inflamatorias y ácidos grasos libres (FFA por sus siglas en inglés), las cuales actuando sobre los tejidos hepático y muscular modificando su respuesta a la inflamación así como el metabolismo lipídico, contribuyendo al síndrome metabólico. En la obesidad se ha demostrado que ocurre una infiltración de macrófagos en el tejido adiposo, los cuales contribuyen de manera importante a la secreción de citoquinas pro-inflamatorias (24) (Figura 4). Aun cuando las citoquinas tienen un efecto fundamentalmente paracrino, entran al torrente sanguíneo y causan inflamación sistémica (22). En la obesidad está bien documentado el incremento de las citoquinas pro-inflamatorias tanto en el tejido adiposo como en la circulación (25).
Figura 4. Inflamación y resistencia a la insulina. Durante la obesidad se produce una inflamación crónica de bajo grado del tejido adiposo, acompañada de infiltración por macrófagos. Tanto los adipocitos como los macrófagos producen citoquinas pro-inflamatorias, las cuales actuando localmente y de forma sistémica condicionan: la fosforilación de IR e IRS en serina/treonina, aumento de la expresión de SOCS3 y PTB 1B, incremento en la producción de ceramidas e inhibición de la síntesis de GLUT4. Los aspectos antes mencionados conducen a la resistencia a la insulina.
Las citoquinas pro-inflamatorias producidas por el tejido adiposo y por los macrófagos incluyen: resistina, factor de necrosis tumoral α (TNFα por sus siglas en inglés), interleuquinas (IL por sus siglas en inglés) 6; 18 y 1β, proteína quimiotáctica de monocitos 1 y angiotensina II (26, 27). Como se mencionó antes, estos factores contribuyen al proceso de inflamación local y sistémico que acompañan a la obesidad, pero además condicionan resistencia a la insulina, en particular TNFα, IL6, 18 y 1β y angiotensina II (22), mediante múltiples mecanismos, tales como: activación de quinasas que fosforilan en restos de serina y treonina a IR e IRS, disminución de la expresión de IRS1, GLUT4 y del receptor de proliferación activado de peroxisomas γ (PPARγ por sus siglas en inglés) y/o la expresión y activación del supresor de la señalización de citoquinas 3 (SOCS3 por sus siglas en inglés) (13).
Otro factor importante a considerar en la inflamación asociada a la obesidad lo constituye la activación de los receptores de tipo toll (TLR por sus siglas en inglés) los cuales son una familia de receptores de membrana asociados a la respuesta inmune innata, son activados por compuestos tales como lipopolisacaridos y que condicionan inflamación mediante la vía del factor nuclear kB (NF-kB por sus siglas en inglés) (13). Una de las isoformas de esta clase de receptores, el LTR4 se incrementa su expresión en músculo y tejido adiposo en la obesidad y es activado por ácidos grasos saturados. Se han descrito varios mecanismos por los cuales la activación de LTR4 condicionan la fosforilación de IRS en restos de serina/treonina, modificando las vías de señalización de la insulina: por la activación de JNK, del inhibidor de la quinasa del factor nuclear k (IKK por sus siglas en inglés) y de MAPK (28). En macrófagos, la vía LTR4/NF-kB es activada por ácidos grasos libres, promoviendo la síntesis de citoquinas tales como IL6; 18 y 1β y TNFα las cuales contribuyen al estado de inflamación del tejido adiposo en la obesidad (29). La activación de LTR4 aumenta la expresión de SOCS3 y de PTP-1B, los cuales son reguladores negativos de las vías de señalización de la insulina condicionando resistencia a la hormona (29). Los ácidos grasos saturados no solo son precursores de las ceramidas, sino que además al activar al LTR4 incrementan la expresión de las enzimas que sintetizan ceramidas, el incremento intracelular de éstos compuestos activan a PP2A la cual defosforila proteínas entre las cuales destaca Akt/PKB inhibiéndola, con lo cual afectan la vía de señalización de la insulina (22, 30). Estos datos que relacionan a LTR4 con la inflamación y la resistencia a la insulina sugieren que el sistema inmunológico juega un importante papel en el desarrollo de la DM2.
En la obesidad, la inflamación del hígado está asociada con esteatosis hepática, aumento de la producción de citoquinas pro-inflamatorias y acumulación de células de Kupffer (macrófagos hepáticos). Aun cuando se conoce que las citoquinas inhiben las vías de señalización de la insulina en hepatocitos, el significado biológico de la inflación, en éste órgano, no está completamente claro. Se ha demostrado que la inflación produce resistencia a la insulina y en consecuencia aumento de la producción hepática de glucosa al promover la neoglucogénesis (22).
Las vías de señalización de la insulina en el tejido muscular no son sensibles a la inflamación (22).
La existencia de una estrecha correlación entre la inflación y la patogénesis de la resistencia a la insulina está bien documentada en la literatura, sin embargo la inflamación no es un buen blanco para el tratamiento de la resistencia a la insulina. El uso de drogas antiinflamatorias no mejora sustancialmente la sensibilidad a la insulina, aun cuando en algunos ensayos se ha reportado disminución de los niveles de glicemia (22). Otros aspectos interesantes a destacar son: el incremento de los niveles plasmáticos de IL6 (citoquina pro-inflamatoria) está asociado con el incremento de la sensibilidad a la insulina durante el ejercicio físico (31); las citoquinas pro-inflamatorias estimulan el consumo energético y reducen la ingesta de alimentos lo cual aumenta la sensibilidad a la insulina (32) y que la inflamación promueve un balance energético negativo lo cual es conocido que incrementa la sensibilidad a la insulina (33).
Estrés del retículo endoplasmático y resistencia a la insulina
El retículo endoplasmático (ER por sus siglas en inglés) es un organelo formado por membranas paralelas que forman sacos aplanados conectados entre sí, el mismo desempeña importantes funciones entre las cuales vale la pena mencionar: síntesis de proteína, síntesis de lípidos y algunos esteroides, modificaciones post-traduccionales de proteínas tales como glicosilación y plegamiento tridimensional, transporte de proteínas al aparato de Golgi, almacenamiento de calcio gracias a la participación de la bomba retículo sarco/endoplamática de Ca++ATPasa (SERCA por sus siglas en inglés) la cual transporta activamente el ión desde el citosol a la cisterna del ER.
Dada su importancia, vale la pena destacar que las chaperonas del ER y las enzimas de plegado son las que participan en la adquisición de la configuración tridimensional de las proteínas en la cisterna del ER, entre estas podemos mencionar: las proteínas reguladas por glucosa (GRP por sus siglas en inglés) 78 y 94, la proteína isomerasa de disulfuro, calnexina y calreticulina; es interesante mencionar que además impiden la agregación de las proteínas desplegadas o mal plegadas(5).
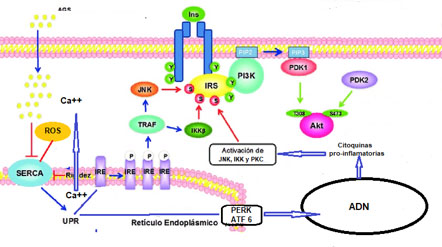
En condiciones de estrés celular(34), como en el caso de la producción de especies reactivas de oxígeno (ROS por sus siglas en inglés) tales como peróxido de hidrógeno y oxígeno singlete, o de un incremento en la concentración de ácidos grasos saturados, se altera la homeostasis del ER; entre otros aspectos vale la pena destacar que la actividad de SERCA se inhibe por efectos de tales compuestos(35), condicionando la depleción de Ca++del organelo (Figura 5), ésta disminución de la concentración del ión en la cisterna de ER determina que las chaperonas dependientes de Ca++que participan en el plegado tridimensional de las proteínas(36), pierdan actividad y en consecuencia se acumulan proteínas.
Figura 5. Estrés del Retículo endoplasmático.En condiciones de estréscelularpor incremento de la concentración de ácidos grasos saturados (AGS) y/o especies reactivas de oxígeno (ROS) se inhibe la actividad de SERCA, condicionando una depleción de Ca++del ER lo cual dispara un mecanismo compensatorio conocido como URP, activándose tres quinasas IRE1, PERK y ATF6. La primera de las quinasa interactuando con TRAF2 activa las proteína quinasas JNK e IKK las cuales fosforilan a IRS en restos de serina/treonina. Por otro lado PERK y ATF6 actuando sobre NK-kB estimulan la expresión de genes que determinan la síntesis de TNFα, IL1β y 6, éstas a su vez activan a JNK, IKK y PKC con las consecuencias antes descritas. La fosforilación de IRS en restos de serina/treonina lo inactivan produciendo resistencia a la insulina.
desplegadas o mal plegadas con lo cual se inhibe su transporte al aparato de Golgi. El conjunto de las alteraciones antes descrito se conoce como el estrés del ER(37). El estrés del ER, activa un mecanismo compensatorio conocido como respuesta a proteínas desplegadas (UPR por sus siglas en inglés) el cual incluye, la inhibición de la síntesis de proteínas así como incremento en la degradación de las mismas en la cisterna de ER y un incremento de las chaperonas del ER y de las enzimas plegadoras en un intento de mejorar el plegado de las proteínas e impedir la agregación de las proteínas desplegadas o mal plegadas(38). El incremento de las proteínas desplegadas o mal plegadas, que ocurre en el UPR, activa tres quinasas sensores del estrés (Figura 5): la proteína quinasa del retículo endoplámatico quinasa similar al ARN (PERK por sus siglas en inglés), la quinasa/endoribonucleasa que requiere inositol 1 (IRE 1 por sus siglas en inglés) y el factor 6 de activación transcripcional (ATF 6 por sus siglas en inglés).
Se ha demostrado una estrecha correlación entre el estrés del ER, la inflamación y la resistencia a la insulina(37); en este sentido vale la pena mencionar que la activación de PERK condiciona la inhibición de la traducción del inhibidor de NF-kB (IkB por sus siglas en inglés) con lo cual se promueve la translocación al núcleo de NK-kB, donde éste estimula la expresión de una variedad de genes involucrados en la inflamación, entre los cuales destacan TNFα, IL1β e IL6, cuyos productos estimulan la actividad de proteína quinasas de serina/treonina tales como JNK, IKK y PKC, estas últimas a su vez fosforilan a IR e IRS1 con lo cual se afecta la vía de señalización de la insulina, como ya se mencionó antes, condicionando resistencia a la hormona(5). Por otro lado IRE1 interactúa con el factor 2 del receptor asociado a TNFα (TRAF2 por sus siglas en inglés) lo cual conduce a la activación de IKK y JNK con las consecuencias arriba descritas. En el caso de ATF2, éste estimula directamente a TNFα con igual resultado que lo mencionado antes.
Es interesante menciona que la existencia de estrés del ER ha sido reportado en hígado y tejido adiposo en humanos con obesidad y que su condición mejora cuando el paciente reducen de peso(39).
Se ha observado que el estrés del ER así como la resistencia a la insulina mejoran en los casos en los cuales se incrementan las expresión de las chaperonas del ER, tales como la proteína chaperonina de 150 KDa o GRP78(40). Así mismo, la administración crónica a humanos obesos, de las chaperonas químicas: 4 fenilbutirato o ácido taurodesoxicolico mejoran la sensibilidad a la insulina y disminuyen el estrés de ER(41, 42).
Disfunción mitocondrial y resistencia a la insulina
Las mitocondrias presentan una estructura doble membranosa y desempeñan un importante papel en el metabolismo energético, en ellas se oxidan los ácidos grasos, por un proceso denominado β oxidación y ocurre la parte final del catabolismo de la glucosa. En ambos procesos se generan coenzimas reducidas, cuyos electrones son transferidos a la cadena respiratoria y en el funcionamiento de la misma se genera la mayor cantidad del ATP celular.
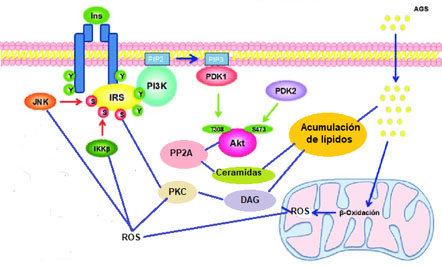
Se define como disfunción mitocondrial, una reducción en el número, densidad o función de las mitocondria (22) trayendo como consecuencia una acumulación de ácidos grasos libres y otros lípidos con la consecuente resistencia a la insulina (43). La acumulación de ácidos grasos libres aumenta los niveles diacilgliceridos (DG) y ceramidas, los primeros activan la PKC con la consecuente fosforilación e inactivación de IR e IRS y las segundas regulan negativamente Akt/PKB afectando la señalización de la insulina (Figura 6).
Por otro lado, existen evidencias experimentales que las especies reactivas de oxígeno (ROS) desempeñan un papel importante en la patogénesis y evolución
Figura 6. Disfunción mitocondrial y resistencia a la insulina. La disfunción mitocondrial causa disminución en la utilización de los ácidos grasos saturados con lo cual ocurre una acumulación de los mismos, como consecuencia se incrementa la síntesis de ceramidas y diacilgliceridos (DAG). Las ceremidas activan a las proteína fosfatasa 2A (PP2A) las cuales defosforilan e inactivan a Akt/PKB inhibiendo la rama metabólica de la señalización de la insulina. Los DAG activan a PKC la cual fosforila, en restos de serina/treonina, e inactiva a IR e IRS. Por otro lado, la disfunción mitocondrial también se manifiesta por incremento en la producción de especies reactivas de oxígeno (ROS) las cuales activan a las proteína quinasa de serina/treonina PKC, IKK y JNK con la consecuente fosforilación e inactivación de IR e IRS. El conjunto de estas alteraciones condiciona resistencia a la insulina.
la DM2 así como en la aparición de las complicaciones de largo plazo. Se ha propuesto que la disminución en la oxidación de los substratos, a nivel mitocondrial, afecta el funcionamiento de la cadena respiratoria con la reacción directa de los transportadores electrónicos con el oxígeno dando origen a radicales superoxido. La producción de ROS condiciona daño a nivel mitocondrial y celular por la oxidación y peroxidación de ADN, proteínas y lípidos, alteraciones que pueden ocasionar mitofagia (eliminación de las mitocondrias dañadas) y apoptosis. La disminución del número de mitocondrias condiciona acumulación de lípidos con las consecuencias antes descritas. Así mismo es conocido que el aumento en la cantidad de ROS activan a las proteínas quinasas de serina/treonina, tales como IKK, JNK y PKC las cuales como se mencionó antes fosforilan e inactivan a IR e IRS produciendo resistencia a la insulina (44). Además se ha reportado que la producción de ROS inhibe la liberación de insulina por las células β pancreáticas.
A pesar de lo antes expuesto, es muy importante destacar que la mayoría de las evidencias existentes sugieren que la disfunción mitocondrial es consecuencia de la resistencia a la insulina (45-47) y no su causa (48). En este contexto cabe mencionar que la insulina estimula la biogénenesis mitocondrial (48); que ni el número ni la función de las mitocondrias varia cuando se incrementa la sensibilidad a la insulina por la reducción de peso en obesos (48). En la obesidad se incremento de la β oxidación de los ácidos grasos, en particular en músculo, hígado y tejido adiposo marrón, como consecuencia de la mayor disponibilidad de los ácidos grasos, aumentando la producción de ATP mitocondrial. El incremento en la concentración de ATP inhibe a la quinasa sensible a adenisina monofosfato (AMPK por sus siglas en inglés), un sensor metabólico que al inhibirse bloque la captación de glucosa por el tejido muscular y adiposo condicionando resistencia a la insulina. En resumen el incremento de la actividad mitocondrial (β oxidación) condiciona resistencia a la insulina (46, 47). Las drogas anti-diabéticas como la metformina y la tiazolidinedionas, entre otras, inhiben la producción de ATP por las mitocondrias mejorando la resistencia a la insulina (50, 51). Las observaciones antes mencionadas sugieren fuertemente que la estimulación de la actividad de las mitocondrias, durante la obesidad, condicionan resistencia a la insulina.
Hiperinsulinismo y resistencia a la insulina
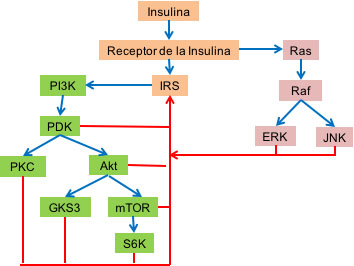
Por hiperinsulinemia se entiende, una elevación constante de los niveles plasmáticos de insulina aun en condiciones de ayuno. Aun cuando generalmente se acepta que la resistencia a la insulina condiciona hiperinsulinismo, muchos estudios sugieren que niveles altos de insulina producen resistencia a la hormona, especialmente si existen niveles altos de ácidos grasos (52). En la vía de señalización de la insulina existe un asa de retroalimentación negativa (Figura 7) la cual es activada por la hormona, en condiciones de hiperinsulinemia, permitiendo la inactivación y/o terminación de la rama metabólica (vía del PI3K, Figura 1) y de la rama mitogénica (vía de Raf/Ras/MEK/ MAPK, Figura 3) de tal vía de señalización; siendo los blancos de dicho control negativo: IR e IRS con lo cual se produce resistencia a la insulina.
Figura 7. Asa de retroalimentación negativa. De la vía de señalización de la insulina se destaca en verde, la rama metabólica (vía del PI3K) y en morado, la rama mitogénica (vía de Raf/Ras/MRK/MAPK) y la secuencia de eventos se muestra por flechas azules. Existe un asa de retroalimentación negativa por la cual, las proteína quinasas de serina/treonina, de la rama metabólica (PDK, PKC, Akt, GKS3, mTOR y S6K) y las de la rama mitogénica (ERK y JNK) fosforilan a IR e IRS inhibiéndolos, la secuencia de eventos de dicha asa se muestran con flechas en rojo. La fosforilación de IR e IRS produce resistencia a la insulina.
La información disponible sugiere fuertemente que la insulina activa proteína quinasas de serina/treonina, entre las cuales están: JNK y ERK, de la vía mitogénica y PDK, Akt/PKB, mTOR, GSK3, PKC y S6K de la vía metabólica; las cuales fosforilan e inactivan a IR e IRS. Esta asa de retroalimentación negativa permite, en condiciones normales, garantizar un adecuado control dinámico de la vía de señalización de la insulina (52).
Conclusiones
En la actualidad se dispone de una amplia información del mecanismo de acción de la insulina, aun cuando faltan algunos aspectos relevantes tales como los relacionados con la estructura y función de IR y con la exocitosis de GLUT4.
Se ha avanzado mucho en el conocimiento del mecanismo molecular de la resistencia a la insulina, sin embargo no se ha logrado establecer aspectos claves de dicho proceso que permitan diseñar fármacos capaces de controlar la resistencia a la hormona y en consecuencia su progresión hacia el desarrollo de DM2.
Referencias
Samuel VT and Shulman GI. 2016. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest. 126: 1222.
De Meyts P. 2016. The Insulin Receptor and Its Signal Transduction Network. De Groot LJ, Chrousos G, Dungan K, et al., editors. South Dartmouth (MA): MDText.com, Inc.; 2000.
Cantley LC. 2002. The phosphoinositide 3-kinase pathway. Science 296:1655-1657.
Thong FSL, Dugani CB and Klip A. 2005. Turning Signals On and Off: GLUT4 Traffic in the Insulin-Signaling Highway. Physiology 20: 271-284.
Avruch J. 2007. MAP kinase pathways: the first twenty years. Biochim Biophys Acta 1773:1150-1160.
Boucher J, Kleinridders A and Kahn CR. 2014. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol. 6(1).pii: a009191.
Olivares-Reyes JA. 2012. Bases moleculares del síndrome metabólico y resistencia a la insulina. En: Garibay Nieto GN, García Velasco S, eds. Obesidad en la edad pediátrica: prevención y tratamiento. Ciudad de México: Corinter; p. 185-214.
Zick Y. 2005. Ser/Thr phosphorylation of IRS proteins: a molecular basis for insulin resistance. Sci STKE. 2005 (268):pe4.
Montagnani M, Ravichandran LV, Chen H, Esposito DL and Quon MJ. 2002. Insulin receptor substrate-1 and phosphoinositide-dependent kinase-1 are required for insulin-stimulated production of nitric oxide in endothelial cells. Mol Endocrinol. 16:1931-1942.
Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R and Zeiher AM. 1999. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 399: 601-605.
Lee JH and Ragolia L. 2006. AKT phosphorylation is essential for insulin-induced relaxation of rat vascular smooth muscle cells. Am J Physiol Cell Physiol. 291: C1355-1365.
Olivares-Reyes JA and Arellano-Plancarte A. 2008. Bases moleculares de las acciones de la insulina. Rev Edu Bioq. 27: 9-18.
Kido Y, Burks DJ, Withers D, Bruning JC, Kahn CR, White MF and Accili D. 2000. Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J Clin Invest. 105: 199-205.
Chavez JA, Knotts TA, Wang LP, Li G, Dobrowsky RT, Florant GL, and Summers SA. 2003. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids.J Biol Chem. 278: 10297-10303.
Ye J. 2013. Mechanisms of insulin resistance in obesity. Front Med. 7: 14-24.
Park HS, Park JY and Yu R. 2005. Relationship of obesity and visceral adiposity with serum concentrations of CRP, TNF-alpha and IL-6. Diabetes Res Clin Pract. 69: 29-35.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL and Ferrante AW Jr. 2003. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 112: 1796-1808.
Halberg N, Wernstedt-Asterholm I and Scherer PE. 2008. The adipocyte as an endocrine cell. Endocrinol Metab Clin North Am. 37: 753768.
Shoelson SE, Lee J and Goldfine AB. 2006. Inflammation and insulin resistance. J Clin Invest. 116: 1793-1801.
Ouchi N, Parker JL, Lugus JJ and Walsh K. 2011. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 11: 85-97.
Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H and Flier JS. 2006. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 116: 3015-3025.
Watanabe Y, Nagai Y and Takatsu K. 2013. Activation and regulation of the pattern recognition receptors in obesity-induced adipose tissue inflammation and insulin resistance. Nutrients. 5: 3757-3778.
Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, Knotts TA, Shui G, Clegg DJ, Wenk MR, Pagliassotti MJ, Scherer PE and Summers SA. 2011. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest. 121: 1858-1870.
Pedersen BK. 2007. IL-6 signaling in exercise and disease. Biochem Soc Trans. 35: 12951297.
Straub RH, Cutolo M, Buttgereit F and Pongratz G. 2010. Energy regulation and neuroendocrine-immune control in chronic inflammatory diseases. J Intern Med. 267: 543560.
Ye J and Keller JN. 2010. Regulation of energy metabolism by inflammation: a feedback response in obesity and calorie restriction. Aging (Albany NY) 2: 361368.
Onyango AN. 2018. Cellular Stresses and Stress Responses in the Pathogenesis of Insulin Resistance. Oxid Med Cell Longev. 2018: 4321714.
Onyango AN. 2017. The Contribution of Singlet Oxygen to Insulin Resistance. Oxid Med Cell Longev. 2017: 8765972.
Braakman I and Hebert DN. 2013. Protein folding in the endoplasmic reticulum. Cold Spring Harb Perspect Biol. 5: a013201.
Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH and Hotamisligil GS. 2004. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 306: 457-461.
Yalcin A and Hotamisligil GS. 2013. Impact of ER protein homeostasis on metabolism. Diabetes. 62: 691-693.
Gregor MF, Yang L, Fabbrini E, Mohammed BS, Eagon JC, Hotamisligil GS and Klein S. 2009. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss.Diabetes. 58: 693-700.
Ozawa K, Miyazaki M, Matsuhisa M, Takano K, Nakatani Y, Hatazaki M, Tamatani T, Yamagata K, Miyagawa J, Kitao Y, Hori O, Yamasaki Y and Ogawa S. 2005. The endoplasmic reticulum chaperone improves insulin resistance in type 2 diabetes. Diabetes. 54: 657-663.
Xiao C, Giacca A and Lewis GF. 2011. Sodium Phenylbutyrate, a Drug With Known Capacity to Reduce Endoplasmic Reticulum Stress, Partially Alleviates Lipid-Induced Insulin Resistance and β-Cell Dysfunction in Humans. Diabetes. 60: 918-924.
Kars M, Yang L, Gregor MF, Mohammed BS, Pietka TA, Finck BN, Patterson BW, Horton JD, Mittendorfer B, Hotamisligil GS and Klein S. 2010. Tauroursodeoxycholic Acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes. 59: 1899-1905.
Lowell BB and Shulman GI. 2005. Mitochondrial dysfunction and type 2 diabetes. Science. 307: 384387.
Montgomery MK and Turner N. 2015. Mitochondrial dysfunction and insulin resistance: an update. Endocrine Connect. 4: R1-R15.
Holloszy JO. 2009. Skeletal muscle mitochondrial deficiency does not mediate insulin resistance. Am J Clin Nutr. 89: 463S466S.
Pagel-Langenickel I, Bao J, Pang L and Sack MN. 2010. The role of mitochondria in the pathophysiology of skeletal muscle insulin resistance. Endocr Rev. 31: 2551.
Muoio DM. 2010. Intramuscular triacylglycerol and insulin resistance: guilty as charged or wrongly accused? Biochim Biophys Acta. 1801: 281288.
Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, Neschen S, White MF, Bilz S, Sono S, Pypaert M and Shulman GI. 2005. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest. 115: 35873593.
Stump CS, Short KR, Bigelow ML, Schimke JM and Nair KS. 2003. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc Natl Acad Sci USA. 100: 799678001.
Brunmair B, Staniek K, Gras F, Scharf N, Althaym A, Clara R, Roden M, Gnaiger E, Nohl H, Waldhäusl W and Fürnsinn C. 2004. Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes 53: 10521059.
González-Mujica F. 2016. Drogas antidiabéticas diferentes de la insulina. Mecanismos de acción. Vitae, Abril-Junio Nº 66. Disponible en: http://vitae.ucv.ve/?module=articulo&rv=124&n=5316
Ye J. 2007. Role of insulin in the pathogenesis of free fatty acid-induced insulin resistance in skeletal muscle. Endocr Metab Immune Disord Drug Targets. 7: 6574.
NOTA:Toda la información que se brinda en este artículo es de cará
cter investigativo y con fines académicos y de actualización para estudiantes y profesionales de la salud. En ningún caso es de carácter general ni sustituye el asesoramiento de un médico. Ante cualquier duda que pueda tener sobre su estado de salud, consulte con su médico o especialista.