Freddy González-Mujica
freddygonzalezmujica@gmail.com
Médico. Cirujano. PhD. en Bioquímica
Sección de Bioquímica Médica, Instituto de Medicina Experimental, Facultad de Medicina, Universidad Central de Venezuela. Caracas. Venezuela
Bioquímica Mecanismo de acción de la insulina. (Revisión) Fecha de recepción: 19/06/2017
Fecha de aceptación:
25/11/2017
La insulina es una hormona polipeptídica, anabolizante que ejerce un efecto pleotrópico, el cual es mediado por un receptor (IR) de membrana con actividad de tirosina quinasa. IR es un tetrámero, α2β2, el cual presenta una estructura modular; un dominio extracelular formado por la totalidad de las subunidades α, las cuales están glicosiladas, y una pequeña proporción de la subunidades β, esta última se continua con un segmento incluido en la membrana plasmática seguido luego por el dominio citoplasmático. En el dominio extracelular se encuentran los sitios de unión a la insulina y en el dominio citoplasmático radica la actividad de tirosina quinasa. Cuando IR se une al ligando se promueve la autofosforilación del receptor y la fosforilación de substratos específicos de IR (IRS por sus siglas en inglés) iniciándose redes de señalización. Se postula la existencia de tres nódulos críticos en la señalización intracelular de la insulina a saber: IR e IRS, la fosfatidilinisitol 3-quinasa (PI3K por sus siglas en inglés) y la proteína quinasa B (AKT/PKB por sus siglas en inglés). Luego de la autofosforilación de IR y la fosforilación de IRS se generan tres diferentes vías de señalización: La de Raf/Ras/MEK/ MAPK, la de PI3K y la alterna que involucra CAP-APS-Cbl-TC10-aPKC; las dos últimas vías están relacionadas con la translocación del transportador de glucosa 4 (GLUT4 por sus siglas en inglés) del citosol a la membrana plasmática con el consecuente incremento de la captación de glucosa por músculo y tejido adiposo. La primera vía está relacionada con la regulación de la expresión genética. También se discute algunos aspectos de la finalización de la señal de la insulina tales como la defosforilación de IR e IRS por tirosina fosfatasas y su fosforilación en serinas/treoninas, defosforilación de otras quinasas fosforiladas en serina/treonina, la hidrolisis del fosfatidilinositol 3 fosfato (PIP3 por sus siglas en inglés) y por último la internalización y degradación de IR e insulina.
Palabras Claves:insulina, receptor de la insulina, substratos del receptor de insulina, fosfatidilinositol 3 quinasa, GLUT4
Title Mechanism of Action of Insulin (Review)
Abstract
Insulin is a polypeptidic, anabolic hormone that exerts a pleotropic effect, which is mediated by a membrane receptor (IR) with tyrosine kinase activity. IR is a tetramer, α2β2, which has a modular structure; an extracellular domain consists of all α subunits, which are glycosylated and a small proportion of β subunits, the latter being continued with a segment included in the plasma membrane followed by the cytoplasmic domain. In the extracellular domain are the sites of insulin binding and in the cytoplasmic domain lies the activity of tyrosine kinase. When IR binds to the ligand, the receptor is auto phosphorylated and it phosphorylates the IR-specific substrates (IRS) initiating signaling networks. The existences of three critical nodules in the intracellular signaling of insulin are postulated, namely: IR and IRS, phosphatidylinositol 3-kinase (PI3K) and protein kinase B (AKT / PKB). After auto phosphorylation of IR and phosphorylation of IRS, three different signaling pathways are generated: The Raf / Ras / MEK / MAPK, the PI3K and the alternative, involving CAP-APS-Cbl-TC10-aPKC, the last two pathways are related to the translocation of the glucose transporter 4 (GLUT4) from the cytosol to the plasma membrane with the consequent increase of glucose uptake by muscle and adipose tissue. The first pathway is related to the regulation of genetic expression. We also discuss some aspects of the end of the insulin signal such as the dephosphorylation of IR and IRS by tyrosine phosphatases and their serine/threonine phosphorylation, dephosphorylation of other serine/threonine phosphorylated kinases, hydrolysis of phosphatidylinositol 3 phosphate (PIP3) and finally the internalization and degradation of IR and insulin.
La insulina es una hormona
polipeptídica anabólica producida por las células β de los islotes
pancreáticos, la cual juega un papel primordial en la regulación del
metabolismo. Aun cuando ha sido visualizada como una hormona relacionada con la
homeostasis del metabolismo de la glucosa, en la actualidad se sabe que ejerce
un amplio efecto pleotrópico. Un sistema de señalización similar al de la
insulina existe en todos los metazoarios y
regula mecanismos tales como la reproducción y las expectativas de vida (1).
El concepto de que la
insulina actúa promoviendo el paso de la glucosa a través de la membrana
plasmática de las células blanco (en lugar de actuar sobre enzimas específicas
del metabolismo intermediario) fue establecido por Levine y colaboradores(2)
quienes demostraron que la insulina incrementa considerablemente la
distribución de la galactosa en los perros eviscerados y nefrectomizados desde
un 46 % a un 75 % del peso corporal, valor el cual es muy cercano al contenido
de agua del organismo. A partir de esos resultados, postularon la siguiente
hipótesis de trabajo: La Insulina actúa sobre la membrana celular de algunos
tejidos (músculo esquelético, entre otros) de
tal manera que se facilita la transferencia de las hexosas (quizás de otras
substancias también) desde el líquido extracelular al interior de la célula. El
destino intracelular de la hexosa depende de la disponibilidad de la célula de
vías metabólicas para su transformación. En el caso de la glucosa, la
deposición de glucógeno y su transformación en grasas depende de la velocidad
de su entrada a las células.
Un avance importante fue el
concepto de que la insulina actúa sobre un receptor de membrana, el cual fue
primero caracterizado por métodos isotópicos y después bioquímicamente,
estableciéndose su estructura subunitario y su naturaleza glicoproteíca. Estos
primeros estudios han sido revisados porDe Meyts(3).
Luego de la demostración (4)
de que asociado al receptor de la insulina existe una actividad de tirosina
quinasa (transferencia del fosfato ϒ del ATP a una tirosina en un substrato
proteico) varios grupos demostraron que el receptor de la insulina tiene
actividad de tirosina quinasa y de que entre sus substratos se encuentra el
receptor en sí mismo. Posteriormente el receptor fue clonado (5,6).
Una vez que la insulina
se une al receptor, activando su autofosforilación, éste recluta una serie de
proteínas con lo cual se propaga corriente abajo la señal. Entre las proteínas
reclutadas se encuentran: las proteínas substratos del receptor de insulina (proteínas
IRS, por sus siglas en inglés), proteínas Shc (proteína codificada por el gen SHC y tiene relación con la apoptosis y
la resistencia a drogas de las células de mamíferos. Su sobre expresión está
asociada a carcinogénesis y metástasis) y las proteínas SH2B1 y 2 (proteínas
adaptadora para varios miembros de la familia de receptores de tirosina
quinasa); las dos primeras (proteínas IRS y Shc)contienen dominios que unen fosfotirosina
(PTB por sus siglas en inglés) y las dos
últimas (SH2B1 y 2) poseen dominios de homología Src (dominios de unión presentes en la
oncoproteína Src). La activación de estas proteínas condiciona la respuesta
metabólica y/o genética de la célula. En las siguientes secciones discutiremos
las características del receptor de insulina (IR por sus siglas en inglés), la
secuencia de señalización que condiciona la respuestas tanto metabólica como
genética. Dado su importancia, destacaremos los aspectos relacionados con la
translocación a la membrana plasmática del transportador de glucosa GLUT 4 a
partir de vesículas intracelulares. Por último discutiremos la finalización de
la acción de la insulina.
Receptor de Insulina
El genoma humano codifica 55
receptores con actividad de tirosina quinasa los cuales se agrupan en 19
subfamilias dependiendo de su estructura y características funcionales, en
general están constituidos por un monómero el cual se dimeriza al unirse al
ligando. La subfamilia del receptor de la insulina comprende: el receptor de la
insulina (IR por sus siglas en inglés), el cual une insulina; el receptor del factor de crecimiento similar
a la insulina, el cual une los factores
de crecimiento similares a la insulina I y II y el receptor huérfano
relacionado con el receptor de la insulina, el cual no posee ligando conocido.
Los miembros de esta subfamilia existen como dímeros aun en ausencia de ligando
(7).
El IR tiene una estructura
modular y es codificado por un gen ubicado en el cromosoma 19p13.2, el mismo
posee 22 exones (secuencias del ADN o preARNm que codifican secuencia de
aminoácidos en la proteína) y 21 intrones (secuencias del ADN o preARNm no codificantes).
El exón 11, que codifica un segmento de 12 aminoácidos, puede ser
alternativamente eliminado (splicing: eliminación de intrones y unión de
exones) generando 2 isoformas del IR, a saber la isoforma A donde no se expresa
el exón 11 y la isoforma B en la cual se expresa el exón 11, las cuales
difieren discretamente en su afinidad por la insulina y su expresión en los
tejidos, la isoforma A se encuentra en los tejidos fetales y la B en el hígado.
Los IR son sintetizados como un proreceptor el cual es procesado por una enzima
proteolítica del tipo de las furinas para formar las subunidades α y β, la
subunidad α es abundantemente glicosilada, se forman los enlaces difulsuro y
adquieren la estructura tridimensional(8). El receptor de insulina
es un heterotetramero constituido por 2 subunidades α y 2 β (α2β2).
Las dos subunidades α son extracelulares y las dos subunidades β comienzan en
la región extracelular, luego atraviesan la membrana plasmática y terminan en
la región intracelular. Las 2 subunidades α están, altamente glicosiladas,
unidas por 2 enlaces disulfuro (probablemente 4) y cada subunidad α se une a
una subunidad β por un enlace disulfuro. Uno de los sitios de unión de la insulina está ubicado en las subunidades
α y el otro en la interacción de las subunidades αβ; la porción citoplasmática
de la subunidad β contiene el dominio de tirosina quinasa (9).
Las subunidades α están
constituidas desde su extremo amino terminal por: un dominio rico en el
aminoácido leucina (L1), seguido por un dominio rico en cisteína (C), luego un
segundo dominio rico en leucina (L2), a continuación un dominio completo de
fibronectina tipo III (F1), seguido de un dominio incompleto de fibronectina
tipo III (F2) para terminar en un largo segmento carboxilo terminal (dominio
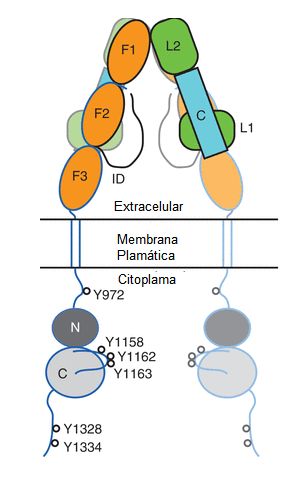
inserto, ID por sus siglas en inglés) (Figura 1)(10).
Figura
1. Estructura esquemática del receptor de Insulina. Las
subunidades α, están ubicadas en la porción extracelular (en el esquema
delineadas en negro) y constituidas por: un dominio rico en leucina (L1) luego
un dominio rico en cisteína (C) seguido por otro dominio rico en leucina (L2) a
continuación esta un dominio completo de fibronectina III (F1) seguido por uno
incompleto de fibronectina III (F2) finalizando en un largo segmento carboxilo
terminal (ID). La subunidad β se encuentra tanto en la parte extracelular como
en el citoplasma y está delineada en azul, comienza con un corto dominio amino
terminal (no se muestra en el esquema), seguido por el complemento del dominio
de fibronectina III F2, a continuación se encuentra un dominio completo de
fibronectina III (F3), luego está el segmento α helicoidal transmembranoso. En
la parte citoplasmática de la subunidad β se encuentran la porción
yuxtamembranosa seguida por el dominio de tirosina quinasa formado por los
lóbulos N y C, en el cual se destaca el asa activadora, por último está en
extremo carboxilo terminal. Se destacan las tirosinas (Y) que son
autofosforiladas por el receptor.
Las subunidades β comienzan
por un corto segmento amino terminal seguido por la porción final del dominio
F2 de fibronectina tipo III seguido por un tercer dominio completo de
fibronectina tipo III (F3) el cual se continua con el segmento de
transmembrana, se piensa que el mismo adopta un configuración α helicoidal. La
porción citoplasmática de la subunidad β está constituida por una porción
yuxtamembranosa de unos 30 aminoácidos seguido del dominio de tirosina quinasa
de unos 300 aminoácidos en el cual se describen dos lóbulos designados N y C,
por último se encuentra la región carboxilo terminal de unos 70 aminoácidos
(Figura 1)(10).
Dado que el receptor comprende
porciones extra e intracelulares, incluyendo un segmento que atraviesa la
membrana plasmática y que posee una gran cantidad de carbohidratos, ha sido
difícil realizar estudios cristalográficos de la molécula completa, en
consecuencia los estudios tridimensionales han estado confinados a los dominios
intra y extracelulares. La estructura del dominio extracelular muestra una
conformación de simetría doble antiparalela (el extremo amino terminal de una
subunidad α se aproxima y empaqueta al extremo carboxilo terminal de la otra
subunidad α) de una V invertida, los dominios F1, F2 y F3 están dirigidas
como un tallo lineal desde la membrana plasmática hacía afuera y los dominios
L1, C y L2 de nuevo hacía la membrana plasmática; así mismo los dominios L1, C
y L2 de un monómero αβ se empaquetan contra las dominios F1, F2 y F3 del otro
monómero(11) (Figura 1).
Se describen dos sitios de
unión del receptor a la insulina: el sitio 1 está formado por el primer dominio
rico en leucina (L1) de una subunidad α y los 16 aminoácidos finales del
extremo carboxilo terminal del dominio ID de la otra subunidad α. Se piensa que
el sitio 2 de unión a la insulina involucra las asas de los dominios de
fibronectina III, F1 y F2 de las subunidades αβ de la otra mitad del receptor; el sitio 1 se considera el más importante de
los sitios de unión de la insulina a su receptor(12).
El análisis cristalográfico
de la porción citoplasmática de la subunidad β ha sido realizado en varios
estados de fosforiloación y en presencia y ausencia de substratos (Mg++
ATP y proteínas). El lóbulo amino terminal N, presenta cinco cadenas de lámina
plegada (configuración β) y una porción α helicoidal; por otro lado el lóbulo
carboxilo terminal C principalmente tiene una estructura α helicoidal (13).
La actividad de tirosina quinasa, del IR, está regulada por el estado de
fosforilación del asa de activación presente en el lóbulo C la cual comienza
con el tripeptido 1150aspartico-fenilalania-glicina
y se extiende hasta la prolina 1170, en la misma se encuentran tres sitios de
autofosforilación, las tirosinas: 1158; 1162 y 1163 las cuales son fosforiladas
en trans cuando el dominio
extracelular se une a la insulina (la unión de la insulina ocurre en un dímero
αβ y la fosforilación ocurre en el otro) (13).
Aun cuando IR es un heterotetrámero
(α2β2) se piensa que el complejo de activación ocurre por
la unión de una sola molécula de insulina asimétricamente al IR con una
afinidad en el orden subnanomolar (0,2 nM), en consecuencia el cambio
conformacional ocurre (o principalmente ocurre) en uno de los dímeros αβ. A
concentraciones mayores de insulina se observa un efecto cooperativo negativo
para la unión a los dos sitios equivalentes en IR(14). Los dos
sitios de unión a la insulina están opuestos cercanamente, sugiriendo que los
cambios conformacionales inducidos por la unión de la hormona al IR sean
relativamente sutiles(15).
En ausencia de insulina,
condición basal, la transfosforilación del dominio de tirosina quinasa está
limitado por un mecanismo desconocido, la unión de la insulina al dominio
extracelular induce una transición estructural en el receptor que facilita la
transfosforilación del dominio citoplasmático de tirosina quinasa. Existen al
menos dos posibles explicaciones para la condición basal: la primera postula
que los dominios de tirosina quinasa están espacialmente separados lo cual
impide el proceso de transfosforilación, en la segunda, los dominios de
tirosina quinasa se encuentran próximos espacialmente pero dispuestos de tal
manera que se impide la transfosforrilación, probablemente por la existencia de
un dímero inhibitorio(16).
En la condición basal y en
la ausencia de Mg++ ATP el asa activadora adopta una configuración
que ocluye el sitio de unión del ATP con lo cual se previene la autofosforilación
de las tirosinas en cis, esto es debido a que la longitud del asa activadora es
tal que no permite la unión simultanea del ATP y la tirosina a fosforilarse
(Y1162) al sitio activo (17).
Se demostró que la primera
tirosina en autofosforilarse, en el asa activadora, es la 1162 seguida por las
tirosinas 1158 y 1163(18). En la estructura cristalina, se ha podido
observar que la tirosina 1162 de una subunidad αβ de IR se encuentra unido al
centro activo de la subunidad αβ contralateral (en trans); esta configuración
se adopta gracias a la fuerte unión del ATP lo cual fuerza al asa activadora a
separarse no ocluyendo el sitio de unión del nucleótido (19).
Una vez que IR es transfosforilado
en las tirosinas 1158; 1162 y 1163, el asa activadora es estabilizada en una
conformación óptima para unir los substratos: Mg++ ATP y las
tirosinas de las proteínas y para que ocurra la catálisis. Aun cuando la
fosforilación de la tirosina 1163 es la más importante para la estabilización
del asa activadora, la fosforilación de las tres tirosinas es importante para
la unión a los substratos del receptor (10).
Además se fosforila una
tirosina en la región yuxtamembranosa (Y 972) la cual es importante para la
unión y activación de las proteínas substratos del IR (IRS por sus siglas en
ingles) y de las proteínas Shc (son 3 proteínas de PM 46; 52 y 66 KDal que
poseen en extremo amino terminal dominios de unión a fosfotirosina y en el
extremo carboxilo terminal dominios SH2). Así mismo se fosforilan 2 tirosinas
(Y1328 y Y1334) en la porción carboxilo terminal (10).
El mecanismo y la cinética
de la unión de la insulina a su receptor ha sido ampliamente estudiado(3),
la misma es compleja y muestra
cooperatividad negativa; solo una molécula de insulina se une al receptor con
alta afinidad y la unión de otra molécula de insulina lo hace con baja afinidad
probablemente por asimetría inducida por ligando.
En la insulina se describen
2 sitios de unión al receptor, el sitio 1 o superficie de unión clásica incluye
aminoácidos de ambas cadenas, siendo los de la cadena A: A1 Gly, A5 Gln, A19
Tyr y A21 Asn y los residuos de la cadena B son: B12 Val, B16 Tyr, B24 Phe, B25 Phe y B26 Tyr. En sitio
2 están incluidos también aminoácidos de ambas cadenas:Ser
A12, Leu A13, Glu A17, His B10, Glu B13 y Glu B17. En general se acepta que el
sitio 1 de la insulina se une al sitio 1 de IR y el sitio 2 de la insulina se
une al sitio 2 del receptor (3).
Aun cuando se ha progresado
bastante en el conocimiento de la estructura de los dominios extracelular y
citoplasmático del IR, en la actualidad no se disponen de estudios
cristalográficos del receptor completo en la presencia y ausencia de
insulina y en consecuencia no se dispone
de información sobre como la unión del ligando al dominio extracelular
condiciona la activación del dominio citoplasmático de tirosina quinasa(7).
Existe controversia sobre el tipo de cambio conformacional que ocurre y si el
incremento de la actividad de tirosina quinasa ocurre por activación o por
eliminación de una actividad inhibitoria.
Transducción intracelular de la señalización de la insulina
Una vez que ocurre la unión
a la insulina al IR el mismo se autofosforila en tres sitios en asa activadora
(Y1158; Y1162 y Y1163) sin embargo las mismas no son responsables por la
interacción con las proteínas con dominios
SH2 (homologías Src 2) o dominios de unión a fosfotirosina (PTB por sus
siglas en inglés) que funcionan como substratos del receptor, por el contrario
las mismas se unen a la tirosina fosforilada Y972 de la región yuxtamembranosa
colocándolas en posición para su fosforilación y activación por el asa
activadora. Estos substratos son: una familia de proteínas substratos del
receptor de la insulina (IRS por sus siglas en inglés) en la cual existen 6
miembros y las proteínas adaptadoras Shc (proteínas con dominios de homología
Src 2)(7) .
Para comprender mejor la
transducción intracelular de la señalización de la insulina es adecuado
postular la existencia de tres nódulos críticos a saber: el IR e IRS, la
fosfatidilinositol 3 quinasa (PI3K por sus siglas en inglés) y la proteína
quinasa B (AKT/PKB por sus siglas en inglés)(20). Las
características del IR fueron descritas en la sección precedente.
De las 6 proteínas IRS las 5
y 6 no tienen actividad de substratos de IR(21), de las cuatro
restantes IRS 1 y 2 son las más importantes y alcanzan una distribución ubicua,
IRS3 se encuentra en los adipocitos y el cerebro mientas que IRS4 se encuentra
en las células embrionarias. IRS 1 y 2 y
las proteínas adaptadoras Shc median efectos metabólicos de la insulina(20).
Los IRS son proteínas que poseen en el extremo amino terminal un dominio de
homología de pleckstrina (es un dominio de unos 120 aminoácidos que existe en
una gama de proteínas relacionadas con la señalización intracelular y el
citoesqueleto y tienen la capacidad de unirse a los fosfolípidos de la membrana
plasmática) adyacente a un dominio de
PTB el cual une a la tirosina fosforilada Y972 de la porción yuxtamembranosa
del dominio citoplasmático de IR. Las regiones centrales y carboxilo terminales
de las proteínas IRS poseen unas 20 tirosinas potencialmente fosforilables por
el IR fosforilado. Luego de activados, por fosforilación, los IRS unen moléculas que contienen dominios
SH2.
Del nódulo IR-IRS parten
dos vías principales de señalización de la insulina, estas son: la vía de la
PI3K (una quinasa de fosfolípidos)/AKT (PKB)(22) y la vía de Raf/Ras/MEK/ MAPK (proteína
quinasa activado por mitógeno también conocida como quinasa regulada por
señales extracelulares, ERK por sus siglas en inglés) (23). La vía
de la PI3K es responsable de la mayoría de los efectos metabólicos de la
insulina y está conectado exclusivamente a IRS, por otro lado, la vía de la
MAPK parte de ambos IRS y Shc y está relacionada con la regulación de la
expresión genética y con la cooperación entre PI3K y el control del crecimiento
(mitogéneis) y diferenciación celular(20).
Vía de la Fosfatidilinositol
3 quinasa.
La activación de la vía de
la PI3K, se inicia con la unión de los 2 dominios de SH2 presentes en la
subunidad regulatoria (p85) de la enzima a los IRS 1 y/o 2 fosforilados por IR,
con lo cual se activa la subunidad catalítica (p110) la cual fosforila al
fosfatidilinositol 4; 5 bisfosfato generando
fosfatidilinositol-3; 4; 5 trifosfato (PIP3), este fosfolípido actúa
como segundo mensajero, se une por medio
de un dominio de homología de pleckstrina a la proteína quinasa dependiente de
fosfoinositol (PDK por sus siglas en inglés) activándola. Por su parte PDK fosforila y
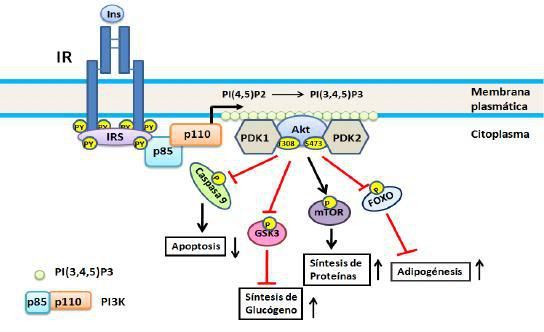
activa a AKT/PBK (20) (Figura 2).
Figura
2. Vía de PI3K. Se
inicia por la unión de la subunidad reguladora (p85) de la PI3K a los IRS
fosforilados con lo cual se activa la subunidad catalítica (110), esta a su vez
fosforila a PIP2 transformándolo en PIP3, este último
actuando como segundo mensajero activa a PDK la cual fosforila a AKT/PBK. En el
esquema se representan cuatro de los substratos de AKT/PBK: caspasa 9, GSK3,
mTOR y Fox0; no se incluye AS160 ya que ésta será objeto de consideración
aparte. Para detalles ver el texto.
AKT/PBK, son una familia de
proteína quinasas de serina/treonina que poseen un dominio de homología de
pleckstrina lo cual permite su reclutamiento a la membrana plasmática para
unirse a PIP3. Los substratos más importantes del nódulo AKT/PBK son:
- El blanco de la rapamicina
en los mamíferos (mTOR por sus siglas en inglés) involucrada en la regulación
de la síntesis de proteínas (24). mTOR es una proteína quinasa de
serina/treonina que actúa como sensor de nutrientes y que funciona como la
subunidad catalítica de 2 complejos estructuralmente distintos mTORC1 y mTORC2.
mTOR estimula la síntesis de proteínas por fosforilación del factor de
iniciación eucariota E4 que une a la proteína 1 y por fosforilación de la
proteína ribosomal p70 S6 quinasa (24).
- La quinasa 3 de la
glucógeno sintasa (GSK3 por sus siglas en inglés) involucrada en la regulación
de la síntesis de glucógeno 25. La GSK3 es una quinasa de
serina/treonina que inhibe por fosforilación a la glucógeno sintasa. La GSK3 es
inhibida por fosforilación por AKT/PKB con lo cual se estimula la síntesis de
glucógeno.
- La familia 0 de los
factores de transcripción cabeza de tenedores (Fox0 por sus siglas en inglés)
y en particular Fox01 el cual está involucrado en la regulación de los genes
gluconegénicos y adipogénicos (26). Fox01 es un factor de transcripción que en
ausencia de insulina se traslada al núcleo y estimula la expresión de genes
tales como el de la fosfoenol piruvato carboxiquinasa, una enzima clave en la
neoglucogenésis, así como el de la ciclina atípica G2 la cual bloquea el ciclo
celular, Fox01 es inhibido por la
insulina y aparentemente juega un papel muy importante en la mitogénesis
inducida por insulina (27). Fox01 es secuestrado en el citoplasma por
la fosforilación por AKT/PKB y en consecuencia no estimula la transcripción de los
genes antes citados.
- La proteína AS160
(substrato de AKT/PKB de 160 KDa) involucrada en el transporte de glucosa(28),
en vista del papel muy importante que juega en la homeostasis de glucosa será
discutido con algún detalle más adelante.
- Fosforila y activa a la
enzima sintasa de óxido nítrico endotelial (eNOS por sus siglas en inglés) la
cual produce la molécula vasodilatadora y anti-inflamatoria óxido nítrico (NO)
estableciéndose una conexión potencial entre la resistencia a la insulina y las
enfermedades cardiovasculares (29).
- Fosforila directamente la proteínas
caspasa 9, inhibiendo su actividad apoptótica у promoviendo por tanto la
supervivencia celular.
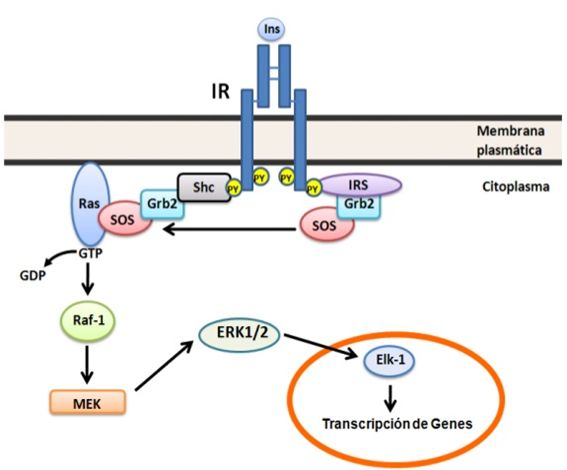
Vía de Raf/Ras/MEK/ MAPK.
Una segunda rama esencial de
la señalización de la insulina la constituye la vía de Raf/Ras/MEK/ MAPK
(Figura 3), la cual es independiente de PI3K y de AKT/PKB. Ambos, IR activado y
las proteínas IRS activadas poseen sitios de unión a proteínas que contienen
dominios SH2 tales como Grb2 (proteína adaptadora que une al factor de
crecimiento 2, está relacionada con la transducción de señales celulares) y Shc
(Son 3 proteínas de PM 46; 52 y 66KDal que poseen en el extremo amino terminal dominios de unión
a fosfotirosina, PBT, y en el extremo
carboxilo terminal dominios SH2). El extremo carboxilo terminal de Grb2 une
proteínas como Gab-1 (es uno de los substratos de las proteínas IRS) y en el
extremo amino terminal une proteínas ricas en prolina tales como hijo de los
sin siete (SOS por sus siglas en inglés) la cual funciona como un
intercambiador de nucleótidos de guanina para Ras (proteínas de bajo peso
molecular que unen nucleótidos de guanina, controlan diferentes fenómenos:
integridad del citoesqueleto; proliferación, diferenciación, adhesión y migración
celular y la apoptosis). SOS permiten la conversión de Ras asociado a la
membrana plasmática de la forma inactiva unido a GDP a la forma activa unido a
GTP. Ras-GTP activa a la proteína quinasa de serina/treonina denominada Raf, la
cual estimula por fosforilación, en serina/treonina, su blanco corriente abajo
MEK1 y 2 (este acrónimo deriva de MAP
quinasa, ERK y Kinasa), estos a su vez
fosforilan (en serina/treonina) y activan a las MAP quinasas (proteína quinasa
activada por mitógeno) la cual activa por fosforilación a ERK1 y 2 (quinasa
regulada por señal extracelular). La activación de ERK1 y 2 juega un papel
directo en la proliferación y diferenciación celular regulando la expresión de
genes y eventos extranucleares tales como la organización del citoesqueleto
mediante la fosforilación y activación de blancos en el núcleo y el citoplasma (23).
Figura 3. Vía de Raf/Ras/MEK/ MAPK. Esta vía se inicia con la interacción de IR e
IRS activados con las proteínas adaptadoras Grb2 y Shc, ésta última une SOS que
actúa como un intercambiador de nucleótidos de guanina en la proteína de bajo
peso molecular Ras. Ras-GTP fosforila y activa a la proteína quinasa Raf, ésta
a vez fosforila a MEK, la cual activa por fosforilación a ERK 1 y 2 que
modifican eventos nucleares y citoplasmáticos. Para detalles ver el texto.
Translocación del GLUT 4 regulado por la insulina.
El efecto clásico de la
acción de la insulita es la entrada de la glucosa a las células del tejido
adiposo, al músculo esquelético y cardiaco(2). La entrada de
glucosa al músculo esquelético es el evento más importante mediado por la
insulina que previene la hiperglicemia postprandial; éste ocurre gracias a la
translocación, desde vesículas intracelulares a la membrana plasmática, del
transportador de glucosa 4 (GLUT4) por un mecanismo que aún no está
completamente claro(30).
El GLUT4 es una de las 13 isoformas de transportadores
de glucosa, los cuales poseen 12 dominios transmembranosos y que se expresa
abundantemente en los tejidos adiposo y muscular. Los GLUTs transportan
monosacáridos por un mecanismo independiente de ATP y sin el requerimiento de
sodio; el GLUT4 tiene la característica distintiva de encontrarse en vesículas
intracelulares (GSVs por sus siglas en inglés) en el estado no estimulado las
cuales son redistribuidas a la membrana plasmática por efectos de la insulina o
por otros estímulos como el ejercicio muscular (31).
En las células adiposas y
musculares, no estimuladas por la insulina, aproximadamente entre un 3-10 % de
los GLUT4 se encuentran en la membrana plasmática y más del 90 % está en
compartimientos intracelulares (GSV). La estimulación por insulina incrementa
considerablemente la exocitosis de GLUT4 a la membrana plasmática reduciendo
mínimamente la endocitosis(32). Concomitantemente ocurre una
redistribución de las GSV, de una forma cónica alrededor del núcleo a la de un
anillo(30). La fusión de las GSV a la membrana plasmática requiere
de unas proteínas de membrana asociadas a vesículas (VAMP2 por sus siglas en
inglés) las cuales pertenecen a la familia de las SNARE (proteínas que median
la fusión de vesículas a sus membranas blancos).
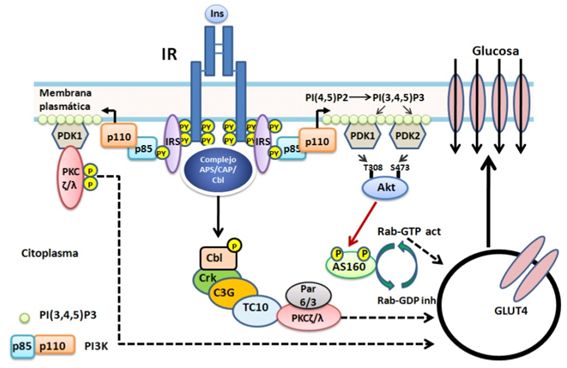
La principal vía involucrada
en la translocación de las GSVs es la PI3K PDK AKT/PBK, (Figura 4) ésta
última fosforila uno de sus substrato AS160 la cual es una proteína que modula la
actividad de GTPasa de la proteína de bajo peso molecular Rab. Cuando AS160 no
está fosforilado y es activa estimula la actividad de GTPasa de Rab generándose
Rab-GDP el cual es inactivo y se bloquea la exocitosis de GLUT4. Por el
contrario la fosforilación de AS160, por AKT/PBK, la inactiva con lo cual
predomina Rab-GTP incrementándose el tráfico de vesículas que contienen GLUT4 a
la membrana plasmática. En mutantes de AS160 se inhibe la translocación estimulada
por insulina de GLUT4(28) lo cual sugiere que funciona como un
regulador negativo que es inhibido por la insulina por medio de AKT/PBK. Así
mismo en ratones knockdown (que no
expresan una proteína) para AS160 se incrementa la cantidad basal de GLUT4 en
la membrana plasmática lo cual es consistente con la función de retención de
GLUT4 en GSVs intracelulares por AS160 que es liberado por la estimulación por
insulina (33).
La proteína quinasa C
atípica (aPKC por sus siglas en inglés) requiere de PIP3 y
fosforilación por PDK pero no está relacionada AKT/PKB; está involucrada en la
translocación de GLUT4 a la membrana plasmática y la captación de glucosa por
los tejidos adiposo y muscular 34. También se ha relacionado con la
regulación de la síntesis de lípidos en el hígado por incremento de la expresión
de la proteína que une el elemento regulador de los esteroles (35).
En los últimos años se ha
descrito una vía independiente de PI3K, que involucra CAP (proteína activada
por catabolíto la cual une AMPc) y APS (proteína adaptadora con dominios de pleckstrina
y SH2 y es substrato del IR) que interactúa con c-Cbl (proteína que une
ubiquitina, está relacionada con la señalización de ubiquitinización y
degradación de proteínas. Posee un dominio que une tirosina fosforilada) en la
translocación de GLUT4 y la captación de glucosa estimulada por la insulina
(Figura 4). Clb es fosforilado en tirosinas por IR y en consecuencia recluta
APS y CAP; posteriormente se une al complejo CrkII (proteínas adaptadoras que
unen varios tipos de proteínas fosforiladas en tirosina además de poseer
dominios SH2), esta a su vez interactúa con un factor liberador de nucleótidos
de guanina (C3G) el cual se comporta como un factor intercambiador de la
proteína de bajo peso molecular que une GTP, TC10 que activaría la aPKC (ζ/λ)
con el consecuente incremento de la translocación de GSV (36).
Finalmente la estimulación de CT10 afecta la dinámica de la actina y la
translocación de GLUT 4 a la membrana plasmática (36).
Se requiere de un
citoesqueleto con una actina dinámica para la translocación de GLUT4 a la
membrana plasmática y el incremento de la captación de glucosa (37),
en las células musculares la remodelación de los filamentos de actina requiere
de PI3K pero no de AKT/PBK(38). Se pudiera postular que la
remodelación de la actina promovida por la insulina provee de una plataforma
física para la migración de las
vesículas que contienen GLUT4 a la membrana plasmática, ayudada por
motores proteicos (39).
Figura
4. Exocitosis de GLUT4. La insulina promueve la translocación de
GLUT4 desde las GSV a la membrana plasmática. La proteína AS160 en su estado no
fosforilado y activo regula negativamente a la proteína G de bajo peso
molecular Rab las cuales participan en el tráfico de GSV. AS160 estimula la
hidrólisis de GTP generando Rab-GDP el cual es inactivo, inhibiendo el tráfico
de vesículas. Cuando AS160 es fosforilado por AKT/PBK se inhibe,
incrementándose Rab-GTP y se incrementa el tráfico de vesículas. Por otra parte
PDK fosforila la aPKC (ζ/λ) la cual incrementa la translocación de GSV. Se ha
postulado una vía alterna que parte de IR el cual activaría a las proteínas
APS-CAP-Cbl y estas activarían a la proteína G de bajo peso molecular TC10 que
activaría aPKC (ζ/λ) la cual incrementa la translocación de GSV.
El ejercicio muscular también
estimula la translocación del GLUT4 a la membrana plasmática través de un
mecanismo no mediado por la insulina y que no depende de PI3K, pero sí de la fosforilación de AS160 por la AMPK
(quinasa dependiente de AMP)(31), la cual funciona como un sensor
metabólico muy importante, se activa alostericamente por aumento de la
concentración intracelular de AMP (disminución concomitante de ATP) y por
fosforilación por quinasas como la proteína quinasa quinasa β dependiente de
calcio/calmodulina (40).
Finalización de la señal y mecanismos de retroalimentación negativos
La activación de proteína quinasas
por la insulina está sujeta a la acción opuesta de proteína fosfatasas y de
mecanismos de retroalimentación negativos, el balance adecuado entre ambos
procesos garantiza una apropiada sensibilidad a la insulina y su alteración
condiciona resistencia a la acción de la hormona. A continuación pasaremos
revista a los mecanismos más importantes de control negativo:
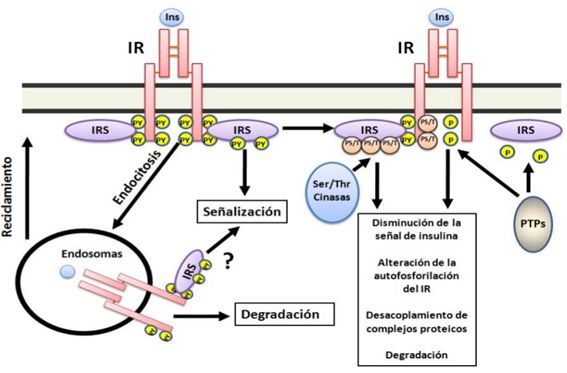
-Fosforilación de serinas en IRS. La
fosforilación en algunas serinas específicas, promovida por la estimulación por
insulina, de IRS condicionan que los mismos no sean reconocidos por IR y/o que
no puedan unirse a la subunidad reguladora de PI3K (p85) o aún más que se
promueva su degradación (41) (Figura 5).
-Fosfatasa
de fosfotirosinas o fosfoserinas/treoninas. La actividad de proteína
fosfatasas que actúen sobre tirosinas fosforiladas como la 1B (PTP1B por sus
siglas en inglés) atenúan la respuesta a la insulina por promover la
defosforilación de IR y de los IRS(42).Otro punto de
control negativo lo constituye la defosforilación e inactivación de AKT/PBK y
aPKC por proteína fosfatasas de serina/treonina 2A(43) (Figura 5).
-Fosfatasas de lípidos. La
fosfatidilinositol-3; 4; 5-trisfosfato 3-fosfatasa (PTEN por sus siglas en
inglés) es una enzima que hidroliza a PIP3 (3; 4; 5) a PIP2 (4;
5) y la fosfatasa de fosfoinositol que contiene dominio SH2 (SHIP2 por sus
siglas en inglés) la cual hidroliza PIP3 (3; 4; 5) a PIP2 (3;
4). La activación de estas enzimas reducen la cantidad de PIP3 con
lo cual se reduce la actividad de AKT/PKB y aPKC disminuyendo los efectos de la
insulina(44).
-Endocitosis del receptor. IR unido a la
insulina es internalizado por endocitosis mediada por clatrina para ser
reciclados o degradado en los lisosomas(45) (Figura 5).
Figura
5. Mecanismos de regulación de la señal de insulina. La
acción de la insulina es modulada por diferentes mecanismos entre los destacan:
la actividad de proteína fosfatasa de tirosina (PTP) que defosforilan residuos
de tirosina tanto en IR como en IRS inactivándolos; fosforilación en
serinas/treoninas en IR y en IRS con lo cual son menos activos y la
internalización de IR unido o no a la insulina para reciclarlo o degradarlo.
En las vesículas
endocíticas, puede ocurrir el inicio de
la degradación de la hormona gracias a la participación de una enzima
específica que degrada insulina (IDE por sus siglas en inglés) o ser
transferida intacta a otros organelos intracelulares como el núcleo, el aparato
de Golgi, el citosol, entre otros (46) o su liberación de la célula
intacta por diacitosis o retroendocitosis (46). La degradación de la
insulina se puede considerar como un mecanismo de terminar su acción.
Conclusiones
Se ha progresado mucho en el
conocimiento de la estructura y el mecanismo de IR; faltan estudios
cristalográficos del receptor en su conjunto y el conocimiento del mecanismo
implicado en la transfosforilación del dominio citoplasmático de IR luego de la
unión a la insulina.
Los nódulos más relevantes
de la red de señalización intracelular de la insulina han sido aclarados, aun
cuando faltan algunos aspectos críticos de los mismos en particular en lo
relacionado con la exocitosis de las vesículas que contienen el GLUT4.
Una mejor comprensión
del mecanismo de acción de la insulina posiblemente facilite el desarrollo de
nuevos fármacos, útiles en el tratamiento de la diabetes, más eficientes y con
menores efectos indeseables.
Referencias
1.White MF. Longevity. Mapping the path to a longer
life. Nature 524:170-171, 2015.
2.Levine R, Goldstein M, Klein S and Huddelston B. 1949.
The action of insulin on the distribution of galactose in eviscerated and
nephrectomized dogs. J Biol Chem 179:985-986.
3.De Meyts P. 2004. Insulin and its receptor: structure,
function and evolution. BioEssays 26:1351-1362.
4.Petruzzelli LM, Ganguly S, Smith CJ, Cobb MH, Rubin CS
and Rosen OM. 1982. Insulin activates a tyrosine-specific protein kinase in
extracts of 3T3-L1 adipocytes and human placenta. Proc Natl Acad Sci USA
79:6792-6796.
5.Ebina Y, Ellis L, Jamagin K, Edery M, Graf L, Clauser
E, Ou J-H, Masiarz F, Kan YW, Goldfine ID, Roth RA and Rutter WJ. 1985. The
human insulin receptor cDNA: the structural basis for hormone-activated
transmembrane signalling. Cell 40:747-758.
6.Ullrich A, Bell JR, Chen EY, Herrera R, Petruzelli LM,
Dull TJ, Gray A, Coussens L, Liao YC, Tsubokawa M, Mason A, Seeburg PH,
Grunfeld C, Rosen M and Ramachandran J. 1985.Human insulin receptor and its
relationship to the tyrosine kinase family of oncogenes. Nature 313:765-761.
7.De Meyts P. 2016. The Insulin Receptor and Its Signal
Transduction Network. De Groot LJ, Chrousos G, Dungan K, et al., editors. South
Dartmouth (MA): MDText.com, Inc.; 2000-.
8.De Meyts P and Whittaker J. 2002 Structural biology of
insulin and IGF1 receptors: implications for drug design. Nat Rev Drug Disc 1:
769-783.
9.Sparrow LG, McKern NM, Gorman JJ, Strike PM, Robinson
CP, Bentley JD, Ward CW. 1997. The disulfide bonds in the C-terminal domains of
the human insulin receptor ectodomain. J Biol Chem 272: 2946029467.
10.Hubbard SR. 2013. The insulin receptor: both a
prototypical and atypical receptor tyrosine kinase. Cold Spring Harb Perspect
Biol. 1; 5 (3) 2013.
11.Smith BJ, Huang K, Kong G, Chan SJ, Nakagawa S,
Menting JG, Hu SQ, Whittaker J, Steiner DF, Katsoyannis PG, Warda CW, Weissb
MA, and Lawrenceal MC. 2010. Structural
resolution of a tandem hormone-binding element in the insulin receptor and its
implications for design of peptide agonists. Proc Natl Acad Sci 107: 67716776.
12.Whittaker L, Hao C, Fu W, Whittaker J. 2008.
High-affinity insulin binding: Insulin interacts with two receptor ligand
binding sites. Biochemistry 47: 1290012909.
13.Hubbard SR. 1997. Crystal structure of the activated
insulin receptor tyrosine kinase in complex with peptide substrate and ATP
analog. EMBO J 16: 55725581.
14.De Meyts P. 2008. The insulin receptor: A prototype
for dimeric, allosteric membrane receptors? Trends Biochem Sci 33: 376384.
15.Ward CW and Lawrence MC. 2012. Similar but different:
Ligand-induced activation of the insulin and epidermal growth factor receptor
families. Curr Opin Struct Biol 22: 360366.
16.Li S, Covino ND, Stein EG, Till JH, Hubbard SR. 2003.
Structural and biochemical evidence for an autoinhibitory role for tyrosine 984
in the juxtamembrane region of the insulin receptor. J Biol Chem 278:
2600726014.
17.Hubbard SR, Wei L, Ellis L, Hendrickson WA. 1994.
Crystal structure of the tyrosine kinase domain of the human insulin receptor.
Nature 372: 746754.
18.Wei L, Hubbard SR, Hendrickson WA, Ellis L. 1995.
Expression, characterization, and crystallization of the catalytic core of the
human insulin receptor protein-tyrosine kinase domain. J Biol Chem 270:
81228130.
19.Wu J, Li W, Craddock BP, Foreman KW, Mulvihill MJ, Ji
QS, Miller WT, Hubbard SR. 2008. Small-molecule inhibition and activation-loop
trans-phosphorylation of the IGF1 receptor. EMBO J 27: 19851994.
20.Taniguchi CM, Emanuelli B and Kahn CR. 2006. Critical
nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell
Biol 7: 85-96.
21.Versteyhe S, Blanquart C, Hampe C, Mahmood S,
Christoff N, De Meyts P, Gray SG and Issad T. 2010. IRS-5 and -6 are poor
substrates for the insulin receptor. Mol Med Rep 3:189-193.
22.Cantley LC. 2002. The phosphoinositide 3-kinase
pathway. Science 296:1655-1657.
23.Avruch J. 2007. MAP kinase pathways: the first twenty
years. Biochim Biophys Acta 1773:1150-1160.
24.Harris TE and Lawrence JC Jr. 2003. TOR signaling.
Sci. STKE 2003, RE 15.
25.Cohen P. 2001. The renaissance of GSK3. Nat Rev Mol
Cell Biol 2:767-776.
26.Accili D and Arden KC. 2004. FoxOs at the crossroads
of cellular metabolism, differentiation and transformation. Cell117:421-426.
27.Svendsen AM, Winge SB, Zimmermann M, Lindvig AB,
Warzecha CB, Sajid W, Horne MC and De Meyts P, 2014. Downregulation of cyclin
G2 by insulin, IGF-I (insulin-like growth factor 1) and X10 (Asp B10
insulin):role in mitogenesis. Biochem J 457:69-77.
28.Sano H, Kane S, Sano E, Miiner CP, Asara JM, Lane WS,
Garner CW and Lienhard GE. 2003. Insulin-stimulated phosphorylation of Rab
GTPase-activating protein regulates GLUT4 translocation. J Biol Chem
278:14599-14602.
29.Yu Q, Gao F and Ma XL 2011. Insulin says NO to
cardiovascular disease. Cardiovasc Res 89: 516524
30.Thong FSL, Dugani CB and Klip A. 2005. Turning Signals
On and Off: GLUT4 Traffic in the Insulin-Signaling Highway. Physiology 20:
271-284.
31.Huang S and Czech MP. . 2007. The GLUT4 glucose
transporter. Cell Metab 5:237-252
32.Satoh S, Nishimura H, Clark AE, Kozka IJ, Vannucci SJ,
Simpson IA, Quon MJ, Cushman SW, and Holman GD. 1993. Use of bismannose
photolabel to elucidate insulin-regulated GLUT4 subcellular trafficking
kinetics in rat adipose cells. Evidence that exocytosis is a critical site of
hormone action. J Biol Chem 268: 1782017829.
33.Eguez L, Lee A, Chavez JA, Miinea CP, Kane S, Lienhard
GE, and McGraw, TE. 2005. Full intracellular retention of GLUT4 requires AS160
Rab GTPase activating protein. Cell Metab; 2: 263272
34.Farese R. V. 2002. Function and dysfunction of aPKC
isoforms for glucose transport in insulin-sensitive and insulin resistant
states. Am. J. Physiol. Endocrinol. Metab. 283, E1E11.
35.Farese R V. Sajan MP. and Standaert ML. 2005. Atypical protein kinase C in insulin action
and insulin resistance. Biochem. Soc. Trans. 33, 350353.
36.Chiang SH, Baumann CA, Kanzaki M, Thurmond DC, Watson
RT, Neudauer CL, Macara IG, Pessin JE, and Saltiel AR. 2001. Insulin-stimulated
GLUT4 translocation requires the CAP-dependent activation of TC10. Nature 410:
944948.
37.Tong P, Khayat ZA, Huang C, Patel N, Ueyama A, and
Klip A. 2001. Insulin-induced cortical actin remodeling promotes GLUT4
insertion at muscle cell membrane ruffles. J Clin Invest 108: 371381.
38.Wang Q, Somwar R, Bilan PJ, Liu Z, Jin J, Woodgett JR,
and Klip A. 1999. Protein kinase B/Akt participates in GLUT4 translocation by
insulin in L6 myotubes. Mol Cell Biol 19: 40084018.
39.Kanzaki M and Pessin JE. 2001. Insulin-stimulated
GLUT4 translocation in adipocytes is dependent upon cortical actin remodeling.
J Biol Chem 276: 4243642444.
41.Gual P, Marchand-Brustel Y, and Tanti JF. 2005.Positive and negative regulation of
insulin signaling through IRS-1 phosphorylation. Biochimie 87: 99109.
42.Galic S, Hauser C, Kahn BB, Haj FG, Neel BG, Tonks NK,
and Tiganis T. 2005. Coordinated regulation of insulin signaling by the protein
tyrosine phosphatases PTP1B and TCPTP. Mol Cell Biol 25: 819829.
43.Ugi
S, Imamura T, Maegawa H, Egawa K, Yoshizaki T, Shi K, Obata T, Ebina Y,
Kashiwagi A, and Olefsky JM. 2004. Protein phosphatase 2A negatively regulates insulins metabolic
signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1
adipocytes. Mol Cell Biol 24: 87788789.
44.Ono H, Katagiri H, Funaki M, Anai M, Inukai K,
Fukushima Y, Sakoda H, Ogihara T, Onishi Y, Fujishiro M, Kikuchi M, Oka Y, and
Asano T. 2001. Regulation of phosphoinositide metabolism, Akt phosphorylation,
and glucose transport by PTEN (phosphatase and tensin homolog deleted on
chromosome 10) in 3T3-L1 adipocytes. Mol Endocrinol 15: 14111422.
46.Duckworth WC, Bennett RG and Hamel FG. 1998. Insulin
degradation: progress and potential. Endocr Rev. 19: 608-624.
NOTA:Toda la información que se brinda en este artículo es de cará
cter investigativo y con fines académicos y de actualización para estudiantes y profesionales de la salud. En ningún caso es de carácter general ni sustituye el asesoramiento de un médico. Ante cualquier duda que pueda tener sobre su estado de salud, consulte con su médico o especialista.
Instituto de Medicina Tropical - Facultad de Medicina - Universidad Central de Venezuela.
Elaborado por el Centro de Análisis de Imágenes Biomédicas Computarizadas CAIBCO, caibco@ucv.ve
Este portal ha sido desarrollado gracias al apoyo del Fonacit