En
una evaluación urinaria de rutina, en un paciente de sexo masculino de 26 años
de edad, se reportó microalbuminuria, con una proteinuria cuantificada de 750
mg/24 horas. Durante la evaluación clínica del paciente, se demostró que en la
infancia presentó acroparestesias; no había historia familiar de dolor neuropático,
nefropatía, cardiopatía o accidente cerebro-vascular (ACV). El examen físico
fue normal; con un peso 72 Kg,
una talla 1,73m, y sin angioqueratomas. Las pruebas de laboratorio reportaron: hemoglobina (Hb) = 14.2 g/dl, ferritina = 120 ng/ml,
depuración de creatinina (Dcr) = 120 ml/min. La función hepática y las proteínas
plasmáticas dentro de valores normales. Las pruebas realizadas de anticuerpos
antinucleares (ANA), anticuerpos anticitoplasma de neutrófilos (ANCA), factor
reumatoideo (RA-test) y velocidad de sedimentación globular (VSG) fueron
normales. Por otra parte, el ecocardiograma, el ecosonograma renal, la
tomografía axial computarizada (TAC) cerebral y la audiometría, todas fueron normales.
Debido a la proteinuria elevada y de causa no precisada, se efectuó una biopsia
renal, la cual mostró una acumulación de glucoesfingolípidos intracitoplasmáticos
tanto en podocitos como en células epiteliales del túbulo contorneado distal
(TCD), las cuales estaban alargadas y vacuoladas. La microscopia electrónica de
la biopsia renal mostró inclusiones osmofílicas en el citoplasma de todas las
células renales (piel de cebollas o con apariencia de cebra) propias de Enfermedad
de Fabry (EF), dicha enfermedad se confirmó debido a una baja actividad de
alfa-galactosidasa A (GAL-A) tanto en leucocitos como en plasma (<1% del
control); y el resultado de la secuenciación de ADN mostró una mutación R310X
en el exón 6, que no se encontró en sus progenitores y parientes (1).
La
enfermedad de Fabry (EF) es una enfermedad ligada al cromosoma X, que afecta el
metabolismo de glucoesfingolípidos debido a la deficiencia de la enzima
lisosomal α-galactosidasa A. Se caracteriza por el depósito de globotriaosilceramida
(GL3) en varios tipos de células; en las células del riñón está presente desde
temprana edad. La enfermedad de Fabry puede causar falla renal, cardiomiopatía,
y enfermedad vascular cerebral, manifestaciones que pueden provocar gran
morbilidad y muerte prematura. La enfermedad renal es casi universal en los
pacientes masculinos, y los signos iniciales son la microalbuminuria y la
proteinuria, que incluso se ha reportado en niños (2). El caso clínico
descrito anteriormente confiere especial importancia a la
proteinuria en rango no nefrótico como único hallazgo de laboratorio, lo que
permite sospechar que existe daño glomerular, siendo la EF una de las causas
probables. Ha sido reportado que en los hombres con EF durante la tercera
década de su vida, se desarrolla azotemia y proteinuria, no obstante antes de
los 20 años de edad pueden ser detectados depósitos renales de GL3 y disfunción
tubular. Es importante señalar que en los pacientes masculinos de más de 50
años de edad, la proteinuria se manifiesta de manera casi universal, y la
progresión hacia la falla renal es tan alta como la observada en los pacientes diabéticos
(3). En el 2009, de acuerdo a datos obtenidos del Registro Fabry
(RF), el 39% de los pacientes tenían una proteinuria mayor o igual a 300
mg/día, y el 22% mostró valores mayores a 1 gr/día.(4) Durante el
año 2010, según los datos de RF, la relación proteína/creatinina (P/C) en orina,
menor de 1g/g fue del 57,2% en los varones y de 59,7% en las hembras, en
Latinoamérica (5) (Tabla 1).
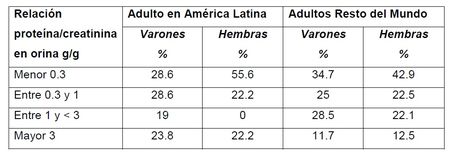 Tabla 1. Relación proteína/creatinina urinaria en pacientes con Enfermedad de Fabry. Datos obtenidos del Registro Fabry (Fuente: Villalobos J, Politei JM et al.(5)
Tabla 1. Relación proteína/creatinina urinaria en pacientes con Enfermedad de Fabry. Datos obtenidos del Registro Fabry (Fuente: Villalobos J, Politei JM et al.(5)
Se
ha reportado con base a los resultados de los
estudios de la fase IV de la terapia de reemplazo enzimática (TRE), que en la EF,
la relación P/C en orina parcial es de fundamental importancia, ya que cuando
la misma es mayor de 1, aumenta la posibilidad que los pacientes desarrollen un
evento Fabry, lo que significa que el paciente puede presentar insuficiencia
cardíaca, cardiopatía isquémica, arritmias y la necesidad de marcapaso,
isquemia cerebral, o el deterioro agudo de la función renal, y la necesidad de
diálisis(6) . Debido a la
incomodidad que representa la recolección de orina de 24 horas para la
cuantificación de la proteinuria, se ha establecido que el cociente P/C muestra
una buena correlación con valores de proteinuria/24 horas comprendidos entre 300-3499 mg. Dicha correlación se mantiene,
pero con menor grado, en valores < 300 mg de proteinuria al día. Cuando la
proteinuria del paciente está en rango nefrótico, el cociente P/C en orina parcial
no se correlaciona con la proteinuria/24 horas.(7) Los valores para comparar P/C con la
proteinuria/24 horas son los siguientes: <0.5 se considera normal, entre
0.5 y 1, se considera dudoso, y >1 se considera anormal.(8)
A partir del año de 1947 se empezaron a estudiar las
lesiones renales características de la EF, y en la década de los años 60,
dichas lesiones se comenzaron a estudiar en material de autopsias, posteriormente en muestras de nefrectomía pre-transplante,
o en biopsias de pacientes con proteinuria y/o falla renal. En 1978, los
investigadores Gubler, Lenoir y col. reportaron
en las biopsias renales de 11 de 12 pacientes con EF (sobre todo en los de sexo
masculino) que en los podocitos, los depósitos lipídicos eran cuantiosísimos
comparado con las células epiteliales tubulares, y casi ausentes en las células
intrínsecas de los glomérulos observados (9) (Tabla 2).
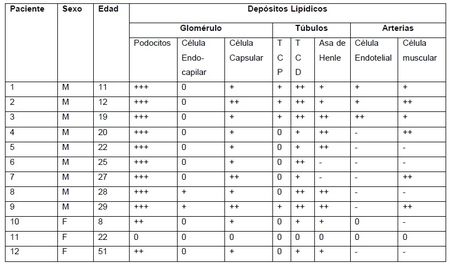 Tabla 2. Datos morfológicos de la microscopía de luz de biobsias renales de pacientes con EF. (Fuente Gubler MC, Lenoir G et al (9) ). TCP: túbulo contorneado proximal. TCD: túbulo contorneado distal. 0 (normal); + (leve); ++ (moderado); +++ (severo); - (no examinado)
Tabla 2. Datos morfológicos de la microscopía de luz de biobsias renales de pacientes con EF. (Fuente Gubler MC, Lenoir G et al (9) ). TCP: túbulo contorneado proximal. TCD: túbulo contorneado distal. 0 (normal); + (leve); ++ (moderado); +++ (severo); - (no examinado)
Los depósitos de
GL3 se observan inicialmente en el glomérulo, y en una primera fase esto se
manifiesta por fusión de los podocitos, y proteinuria leve, acompañada ocasionalmente con microhematuria. La
manifestación a nivel tubular suele ser
posterior a la glomerular, pero clínicamente el defecto de concentración renal
suele surgir antes de la proteinuria. El mecanismo por el cual se produce el
daño renal, específicamente de glomeruloesclerosis, es debido a cambios
isquémicos de la microvasculatura renal, lesión de los podocitos por acumulación de GL3 y por la hiperfiltración.
En las fases avanzadas del daño renal, se observa que hay una sustitución de
las células glomerulares y tubulares por GL3, así como esclerosis glomerular y
depósito de GL3 en el intersticio, lo cual condiciona atrofia tubular
progresiva y fibrosis intersticial. El daño de las estructuras renales no es
uniforme y pueden observarse glomérulos
y áreas intersticiales preservadas (10).
En el año 2013, en
un estudio llevado a cabo por Tondel, Bostad y col, también observaron en biopsias renales
de 12 pacientes con EF, en edades entre 7 y 33 años, que los podocitos y las
células epiteliales del túbulo distal, eran los componentes de la nefrona que
mayor compromiso estructural presentaban aún después de cinco años de TRE (10/12
pacientes, 83,33%), y se observaron inclusiones de GL3 en las células del
endotelio glomerular en 8 de los 12 pacientes evaluados (66,66%); estos
hallazgos llamaron poderosamente la atención, ya que hasta los actuales momentos
la EF ha sido considerada como una enfermedad endotelial (2). Según
Tondel, Bostad y col la importancia clínica del daño podocitario
en la EF fue aclarado por Najafian y col. en el 2011 cuando evaluaron 14
pacientes con EF en edades promedio de 12 años y observaron correlación entre
la proteinuria y los depósitos de GL3, así como daño de los pie de podocitos,
pero no observó correlación entre la proteinuria y el aumento de las
fenestraciones endoteliales (2), inclusive observaron correlación entre
la edad y la cantidad de inclusiones podocitarias. Tondel y col, plantean que existe
una relación entre las inclusiones de GL3 y la disfunción celular, ya que ellos
observaron aclaramiento marcado de las inclusiones podocitarias, así como una
reducción en la fusión de los podocitos y la microalbuminuria de los pacientes
que recibieron TRE para EF durante 5 años.(2) Por todo lo
anteriormente señalado es importante recordar cómo es la fisiología del
podocito.