Asthma and chronic obstructive pulmonary
disease (COPD) comprise chronic inflammatory disorders characterized by airway
hyperresponsiveness and airflow obstruction that can fluctuate over time. They
have an increasing economic burden, not to mention their associated
disabilities and fatal outcomes. Despite of outnumbered research, we still have
not completely understood the big picture of their natural history. Quite a lot
of evidence arising from epidemiological, clinical, pathological, and molecular
studies, have only provided a short view of the pathobiological mechanisms generating
those diseases. The development of better functional assessment techniques
along with a wider availability of new biomarkers allowed to recognize that
inflammatory airway diseases, especially asthma, involve multiple subphenotypes
that differ in clinical severity, histopathology, response to therapy, and
long-term outcome. In consequence, heterogeneous groups have been identified, which
are likely originated from a unique genetic background/ environment
combination.
Severe phenotypes of airway diseases
that course with airway hyperreactivity, implicate airflow obstruction that is
either irreversible or only partially reversible. Their severe nature apparently
is due to longstanding inflammation in the airways. In this setting, injury cyclicity,
age, genetic factors, and previous tissue history induce structural changes; a
phenomenon commonly coined as airway remodeling. Airway remodeling is assumed
to result in severe phenotypes. However, several clinical and animal studies
indicate that the relationship between inflammation, remodeling, and
hyperresponsiveness is complex, and still not completely understood. Considering
that airway hyperreactivity results in an abnormal airway tone, smooth muscle
thickening is thought as the main substrate of abnormal airway mechanics. This
has been deeply explored; hence, considerable data is available. In this
review, we gathered the last experimental findings in this field to formulate a
model of airway smooth muscle (ASM) remodeling that fits into the natural
history of common airway diseases, mainly focusing on molecular mechanisms.
I. Overview of airway smooth muscle remodeling
Growing evidence
supports various pathophysiological mechanisms of airway diseases, including
structural changes seen on severe asthma, COPD, chronic bronchitis, cystic
fibrosis, and bronchiectasis(1-4).
Thus, airway remodeling has been defined by modifications in the composition,
amount, and organization of local cells, including epithelium, glands, blood
vessels, extracellular matrix (ECM), and smooth muscle (see Fig. 1).
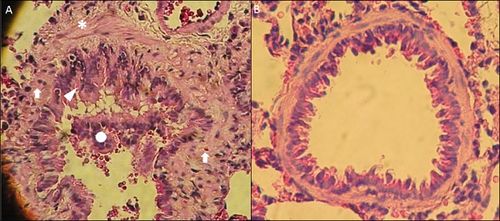
Figure
1. Histopathology of small airways in OVA-sensitized Rats. A
bronchiole from OVA-sensitized rat(A) in comparison to a normal bronchiole from
saline-nebulized rat(B). Tissue shows remodeling features, such as: epithelial
hyperplasia (arrowhead), ASM thickening (asterisk), lymphocytic/eosinophilic
inflammation (arrows), and luminal exudate (circle), compare with the normal
airway. Lung samples were extracted from rats after a protocol previously described(66). Magnification
200X, Hematoxylin-eosin staining.
Interaction of genetic and
environmental factors evolve into assorted outcomes after injury. Many models(5) and clinical studies have shown that
symptoms and functional findings are caused by three interconnected factors: 1)
chronic inflammation, 2) airway hyperresponsiveness (AHR), and 3) tissue
remodeling. Notwithstanding, a major concern in this field is that there is no
clear chronologic and quantitative relationships. A current perspective
considers that unbalanced immune responses to external factors, such as allergens
in asthma, sets up a harmful microenvironment of cyclic injury and repair, which
leads to abnormal structure and function(6,
7). Furthermore, a recent study suggests that chronic mechanical
stress resulting from bronchoconstriction per
se may also lead to remodeling without inflammation(8). Despite of airway remodeling could
be disorder-specific, the airway structure may play a common role to airway
narrowing and airflow limitation carrying out poorly reversible airway
obstruction.
Evidence
of ASM thickening in Asthma and COPD
Asthma and COPD are
well-differentiated clinical entities with some overlapping syndromes in the
middle(3, 9). They share some
features, especially at pathological level, including: epithelial hyperplasia and
dysfunction, subepithelial fibrosis, increased myofibroblasts, increased
vascularization, abnormal neurite branching, and dense ASM layers(2, 7, 10); highlighting that a greater
basement membrane thickness as well as smooth muscle hyperplasia are commonest
seen on asthma(10, 11). Their
deepness frequently wedges with clinical expression and severity(12, 13). A study that evaluated bronchial
wall thickness by high resolution computed tomography in mild-to-moderate
asthma and COPD revealed the airway diameter and thickness were similar(14), but asthma still has received more
attention respect to ASM remodeling. Increased ASM mass could be attributable
to hyperplasia, hypertrophy or both. Under physiological conditions, ASM
located in the central and peripheral airways are bands that wrap up around the
airways in a helical pattern. Its thickness, relative to the diameter of the
airway lumen, increases towards the periphery, but in absolute terms, the
amount is less in the peripheral airways(15).
A morphometric study indicates that the bronchial smooth muscle mass of
patients suffering of fatal asthma was twice than non-asthmatics(16). A major concern of this proposal is
the ECM volume was not measured. To solve it, a recent study showed ASM
hypertrophy in the large airways in both nonfatal and fatal asthma, but
hyperplasia was only seen in large and small airways in fatal cases. Both groups
were associated with an absolute increase in ECM(17). Some degree of airway wall thickening was regularly
detected in asthmatics of all severities with predominance in severe cases(18). The occurrence of remodeling does
not seem to depend on the inflammatory response subtype; since, airway
structure does not differ between asthmatics with eosinophilia and those
without(19). Contrasting
results debate the importance of ASM remodeling because it is not always found
in asthma, hence, no differences in averageof smooth musclecell
cross-sectional area(20).
There is less evidence
supporting ASM thickening in COPD than asthma. Obliteration and fibrosis of the
alveolar wall, mucous gland hypertrophy, and goblet cell hyperplasia are well-known
pathological features of COPD(21).
Increased ASM thickness has been found as compared to control, but lesser than
asthmatics(15). Functional
implications have been shown, as it correlates with the airway obstruction
degree(22). Additionally, ASM
mass and adventitia increased together by 50% in severe COPD affecting the small
airway physiology(23). Biopsy
studies from large airways reported no increase in ASM; moreover, smooth muscle
protein isoforms were not increased, but there was a slight increment in myosin
light chain kinase (MLCK) without changing the myosin light chain
phosphorylation(24). Conflicting
results showed that remodeling may occur in the central airways by greater ECM
protein deposition and increased ASM(25).
This data points out
that asthma and COPD could progress with variable degree of ASM remodeling, but
no direct evidence has been obtained supporting reversibility. A murine model
of asthma suggests that after allergen cessation, the goblet hyperplasia and
collagen deposition resolved first and then lymphocytic infiltration along with
ASM thickening(26). This
brings up an open question whether in human diseases a complete removal of the tissue
hazard can be accompanied by spontaneous resolution of airway remodeling.
Inflammatory
Microenvironment Orchestrates ASM Remodeling
Airway remodeling is associated
with longstanding inflammation. Interleukin (IL)-1β, IL-6, and Tumor Necrosis Factor-α
(TNF-α) as well as growth factors such as Platelet-Derived Growth Factor
(PDGF), Epidermal Growth Factor (EGF), Insulin-like Growth Factor (IGF), and Transforming
Growth Factor-β (TGF-β), have pleiotropic effects; however, specific immune
responses portray distinctive pathologic features(7, 27). T cell reactions cover a wide spectrum of divergent
cytokine networks. In asthma, for example, clinical phenotypes match inflammatory
profiles: I-type hypersensitivity reaction (IgE-dependent), Th2 predominant
inflammation, and non-Th2 associated response(28). However, a common feature seen in all cases is the ASM thickening
(see Fig. 2).
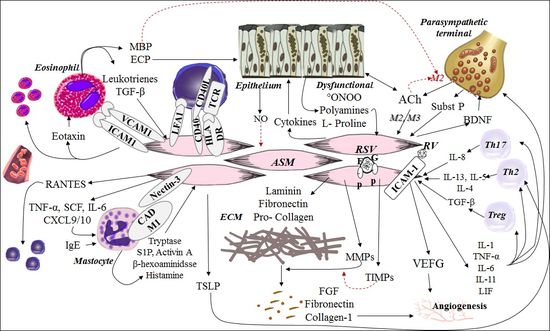
Figure
2. Crosstalk between ASM with cellular and non-cellular components of the
airways during inflammation. (See the text for explanation).
Eosinophils are the
most prominent inflammatory cells in the airways of asthmatics(21). In the course of hypersensitivity
reactions, eosinophils localize in close relation to ASM. For instance, small
airways contain eosinophils in their outer portion (between ASM and alveolar
attachment), whereas in the large airways they are present predominantly in their
inner portion (between ASM and basement membrane)(29). This eosinophil-ASM relationship can enhance cell
proliferation by cysteinyl leukotriene secretion(30). Eosinophil homing in the airways depends on Th2 cytokines
and eotaxin. Adhesion to ASM is mediated by cell adhesion molecules (ICAM-1,
VCAM-1) that are constitutively expressed and also upregulated. ASM cells (ASMC)-derived
cytokines could promote eosinophil differentiation, perpetuating the burden of eosinophils
into ASM bundles(31).
In a similar way, mastocyte
infiltration is prominent in ASM bundles(32).
Mastocyte migration and adhesion potentiate tissue remodeling because histamine,
tryptase, activin A, sphingosine 1-phosphate (S1P), β- hexosaminidase, and
TNF-α, stimulate many ASMC functions(33).
Despite these mediators can stimulate cell proliferation, mast cell seems not
to be relevant for neither proliferation or survival(34). On the other hand, mastocyte can
induce the thymic stromal lymphopoietin (TSLP) in ASM that is highly determinant
of Th2 polarization(35). Mast
cell placement, proliferation and survival into the ASM could occur through allergen-independent
mechanisms(36).
Neutrophilic
inflammation also occurs in severe asthma and COPD. Even though, clinical
phenotypes that course with predominant eosinophilic inflammation lead to ASM
thickening in both large and small airways, the neutrophil infiltration is
almost restricted to concurrent small airway remodeling(37). A histopathological study of
children with fatal untreated respiratory syncytial virus (RSV) infection
showed vascular leakage and neutrophil recruitment into the submuscular layer,
smooth muscle, and airway epithelium, resembling fatal cases of obstructive
diseases(38). IL-8 has been
implicated, and it seems to be secreted by Th17 cells(39). Nowadays, it is known that Th9,
Th22, and Th25 cells also modulate countless aspects of airway immunity(40). Nonetheless, the most significant
cytokine expression in asthma includes IL-4, IL-9, IL-13, eotaxin and RANTES.
This profile correspond to an upregulated Th2 reaction(41). CD4+ T cells transfer
from ovalbumin (OVA)-sensitized rats to non-sensitized rats (adoptive
lymphocyte transfer), showed that an specific subset of Th2 cells drove airway remodeling
in non-sensitized rats after few OVA challenges(42). However, evidence from animal models and humans
indicate that Th2 hypothesis is an incomplete explanation for asthma
pathogenesis, as allergic and nonallergic types are pathologically
indistinguishable(43). It has
also been reported that airway epithelial cells in asthmatics upregulate the EGF
receptor (EGFR) expression, a receptor tyrosine kinase (RTK), even in absence
of significant eosinophilic inflammation(44).
A recent study demonstrated subepithelial fibrosis in severe asthmatics without
evidence of Th2 inflammation(45).
Depletion of CD4+ cells, previous to chronic OVA challenge,
significantly reduced peribronchial inflammation but did not completely reverse
ASM thickening(46). Although
Th2 cytokines have pro-remodeling actions in
vitro, controversial results have been found as IL-5 and IL-13 do not
increase ASMC proliferation, but they induce phenotypic switching(47, 48); and IL-4 inhibits ASMC
replication(49). These studies
suggest that remodeling can also occur independently of Th2 inflammation and
other factors are needed.
COPD is also accompanied
by airway inflammation that is different from asthma, but ASM remodeling still
occur(21). Chronic
inflammation induced by chronic cigarette smoking consists of neutrophil,
macrophage, B cell, and CD8+ T cell recruitment, and it worsens as
disease severity increases(50).
T‐lymphocytes
and macrophages are the predominant cells, being CD8+ T‐lymphocyte
infiltration the most remarkable feature in both large and small airways, and there
is also absence of significant eosinophilic inflammation(51). Although, Foxp3+
regulatory T cells play a role in fibrogenesis, there is no predominant T CD4+
subset. This supports the concept that cyclic events of cytokine and growth
factor surges could be the main drivers regardless of the etiology and immune
polarization.
An intricate network
underlies the ASM and its surroundings, not only immune cells but also neural
parasympathetic endings, mesenchymal cells, ECM, and epithelium (see Fig.2). For example, ASM activation by
proinflammatory cytokines and substance P can induce the brain-derived
neurotrophic factor (BDNF) expression for spatial coordination of neuronal
branching(52), and vascular
endothelial growth factor (VEGF) for control of angiogenesis to assure adequate
perfusion, the latter have an important repercussion on vascular leakage and
vasogenic edema during fatal asthma(53).
Neuronal development also would coordinate spatial distribution of ASM, because
substance P induces both migration and proliferation(54). Nevertheless, the airway epithelium
could have a greater contribution due to its plasticity and inflammatory
properties. Dysfunctional epithelial cells release growth factors, as well as,
acetylcholine (ACh) and leukotrienes that could contribute to ASM growth, ECM
deposition, and angiogenesis(55).
Moreover, the epithelium is an important source of nitric oxide (NO) in the
airways, which has relaxing and other anti-remodeling effects. Physiological NO
is produced by constitutively expressed neuronal
and endothelial NO synthase (n-,e-NOS)(56).
However, cytokines increase inducible NOS (iNOS) and arginase expression. A
greater iNOS/ arginase activity decreases L-arginine bioavailability, which
generates an uncoupled iNOS that not only synthases NO, but it also produces superoxide
and peroxynitrite. These molecules are capable of causing cellular toxicity and
promoting AHR(57). Functional
consequences of increased arginase are reinforced by L-arginine transport blockage
with eosinophil-derived polycations. L-Ornithine, a product of urea cycle, is a
precursor of polyamines and L-proline, both involved in cell proliferation,
collagen synthesis and chromatin remodeling(58).
This exemplifies how noncontractile ASM functions are modulated by many conditions;
therefore, the commonest experimental approaches based on univariate analysis
can under- or overestimate their contribution on smooth muscle processes.
ASMC
are multifunctional
The relevance of ASMCs
in pulmonary diseases has been recognized since the last century. The consensus
until a few years ago was to consider them just as effectors. However, far from
their abilities to contract and relax, ASMCs proliferate, migrate, secrete
chemokines/cytokines, and express surface receptors for cell adhesion and
leukocyte activation, having a crucial role in airway dysfunction(59). A concept of plasticity emerged when
those functions were associated with specific circumstances and required wide adjustment
in gene expression(60, 61). ASM
hypertrophy and/or hyperplasia involve not only outer cell influences, but also
ASMC reactions with paracrine/autocrine properties(7, 62). Quite a lot of molecules could coordinate this loop,
such as: growth factors, cytokines, chemokines, ECM molecules, G
protein-coupled receptor (GPCR) agonists, natriuretic peptides (NPs), NO, and
others(63-66).
Several in vitro synthetic functions have been shown. Also, ASM in mild
asthmatics has constitutive staining for RANTES(67). Further cytokines secreted by ASM include IL-1β and
IL-6 family cytokines, such as leukemia inhibitory factor (LIF) and IL-11(68). These have deeper effects on
recruitment, proliferation, and differentiation of eosinophils, mastocytes, T
cells, and B cells, establishing a bidirectional regulatory network. Mainly, a
CD4+ T cell- myocyte crosstalk through direct contact has shown to be
determinant of ASM remodeling(42).
Airway homing of T cells is CCL5 or RANTES-guided, which is released by ASMCs.
Likewise, strong adhesion between these two cell types has also been described(69). Remarkably, even though ASMCs are
not usually thought as antigen-presenting cells, evidence supports the expression
of major histocompatibility complex class (MHC) II molecules making them capable
of antigen presentation. Moreover, ASMCs express the cell adhesion molecules
(CAMs)/costimulatory molecules, CD40, CD40L, CD80, CD86, ICAM-1 (CD54), VCAM-1 and
LFA-1 (CD11a/CD18)(70). The
CD44-dependent T cell adhesion to ASMCs is not only significant to exchange
inflammatory signals, but also to induce ASM hyperplasia through RTK activation(71). This cooperative signaling mediates
proasthmatic-like changes in ASM responsiveness, and denotes a potential
mechanism of remodeling.
In the airway, a net of
collagenous and noncollagenous proteins influences cellular behaviors. ECM components
include collagens, fibronectin, members of the matrix metalloproteinase (MMP)
family, as well as their inhibitors (TIMP)(59).
After serum stimulation, ASMCs were found to generate elastin, laminin-β1,-2, and
-γ1, thrombospondin, collagen-I-V, and decorin(72). In addition to promoting ECM deposition, ASMCs are
capable to affect its degradation. Human ASMCs release progelatinase A (MMP-2
precursor) and, after TNF-α stimulation, gelatinase B (MMP-9)(73). MMP production suggests that ASM
contributes to ECM turnover, and subsequently the airway remodeling, because inhibition
of the autocrine-derived MMP-2 has antiproliferative effects on ASMC culture(74). Therefore, ECM degradation could be
essential for ASM phenotypic modulation, being degradation of the pericellular
collagen fibrils a requirement to allow cell division(75). Serum levels of TIMP-1 and MMP-9
are raised in both asthma and COPD, supporting a straight relationship between
clinical expression and tissue remodeling. The MMP-9/TIMP-1 ratio and periostin
levels could be consider biomarkers of active disease(76). Cyclic inflammation/ repair simultaneously
occur to cyclic ECM degradation and deposition, which could be a critical phase
in ASM thickening.
Crosstalk
between ASM Remodeling and Hyperresponsiveness
Airway narrowing and
abnormal muscle relaxation are the hallmarks of asthma, COPD, and bronchitis.
Multiple mechanisms have been proposed to explain the AHR, like increased vagal
tone, cytokine-potentiated increment of free intracellular calcium, increased
MLCK activity, and activation of the procontractile Rho kinase pathway(77). All of them have in common that
could hasten the shortening velocity. Therefore, even though remodeling can be triggered
by hypersensitivity reactions, infections, environmental pollutants, and
developmental abnormalities, AHR could be just generated by unbalanced
responses to contractile vs relaxing factors(78). The role of ASM remodeling as a substrate of AHR was uncertain
because functional abnormalities can be seen without changes in the bronchial
smooth muscle mass(79). However,
increasingly data supports a role in severe AHR phenotypes, and irreversible or
partially reversible airflow obstruction(43).
The structure determines both passive tone and active responses to agonist
stimulation. ASM remodeling involves phenotypic changes that enhance its
thickening, and during this process a decline in force induced by repetitive
length changes is seen, but then it rapidly adapts and recovers its ability to
generate force. In this way, higher passive stiffness could contribute to
increased AHR by attenuating the extent of ASM length fluctuations during tidal
breathing, i.e., ASMCs adapt by assuming a shorter resting length while
retaining its ability to generate force(27).
For that reason, after induced bronchoconstriction, deep inspiration causes
airways of asthmatic individuals to dilate transiently.
Expression of
immunomodulatory molecules by ASMCs can delay inflammation resolution and lead
to aberrant healing, which is a potential mechanism of AHR(78). The change in the ASMC population compromises
an increase of synthetic properties, which can modulate the contractile mass. Particularly,
if it is considered that the whole ASM is coupled by gap junctions, and the
calcium dynamic differs between ASMC phenotypes. Propagation of wave-like
calcium currents from modulated ASMC to contractile ASM would hypothetically
affect not only contractile functions but also noncontractile activities, as
discussed in following sections. Other noncontractile elements, including excessive
ECM content, may lead to nonreversible airway obstruction by reducing airway
distensibility(80). Whether increased ASM supports abnormal
reactions to agonists or makes the airway stiffer, it definitely provides an exceptional
substrate for AHR in a framework of progression and severity, at least for
asthma. In fatal asthma, airway wall thickness is increased around 50-230%,
while in nonfatal asthma it ranges from 25 to 150%, most studies pointing out
hyperplasia over hypertrophy as the predominant mechanism(11-13, 43).