Microbiología
Úlceras crónicas: modelo de integración entre patología vascular, inmunológica e infecciosa
Úlceras infecciosas
Las
úlceras infecciosas, son aquellas que son causadas por la
colonización de un microorganismo en una herida causando
debilitamiento de la piel y otros tejidos (subcutáneos) y llegando a
causar fibrosis en la lesión.(1) Úlceras
por micobacterias
Existen
numerosos agentes infecciosos productores de lesiones ulcerosas,
entre los cuales se incluyen
grupos A y G de Estreptococos,
y Pseudomonas
sp., como responsables de piodermas y sepsis. El Staphylococcus
aureus
y la Pseudomonas aeruginosa
pueden estar asociados al
retraso de cicatrización del injerto.(8)
Para
nuestra revisión y posterior elaboración de un modelo de
integración de la génesis de las úlceras crónicas hemos decidido
enfocarnos en una micobacteria,
la cual presentaremos a continuación. A este grupo
se les denomina en ocasiones micobacterias oportunistas o
microorganismos patógenos ocasionales. La mayoría de las especies
pertenecientes a este grupo son casi ubicuas en la naturaleza, y
pueden transformarse en patógenas en circunstancias especiales.
El
Mycobacterium ulcerans, es
un bacilo ácido resistente (BAR) o Ziehl-Neelsen positivo, que crece
muy lentamente en los cultivos entre 6 a 12 semanas y puede tardar
hasta 9 meses en formar colonias visibles en medio sólido.(18) Este
microorganismo es conocido por causar una enfermedad conocida como
úlcera de Buruli; a continuación se presentará una reseña
histórica de la evolución de esta patología.
Se
sabe que después de la aparición de la tuberculosis y la lepra
causadas por dos micobacterias diferentes, apareció una tercera
infección micobacteriana por orden de frecuencia entre los
individuos inmunocompetentes producida por la Mycobacterium
ulcerans. Fue MacCallum quien
describió por vez primera el microorganismo causante al descubrir
bacilos acidorresistentes en una biopsia tomada de una úlcera de la
pierna de un niño en Bairnsdale (Australia) en 1940 y quien publicó
la primera descripción clínica de esta nueva infección
micobacteriana en1948. Sin embargo, la enfermedad ya se conocía en
África antes de 1948 debido a que Sir Robert Cook había descrito en
1897 unas úlceras extensas, causadas casi con toda seguridad por el
Mycobacterium ulcerans.
Entre 1923 y 1935, Kleinschmidt, médico misionero en el nordeste del
Congo, observó también lesiones cutáneas con bordes excavados y
ricas en bacilos acidorresistentes.(19) Posteriormente
a este descubrimiento se fue investigando la etiopatogenia de esta
enfermedad y se observó que la
infección por M. ulcerans se
transmite por medio del ambiente, a través de pequeñas soluciones
de continuidad (heridas) de la piel. Actuando como puerta de entrada
a través de la cual dicha bacteria puede invadir la dermis y el
tejido celular subcutáneo. Un ejemplo: es la transmisión durante
los trabajos de campo, por medio de su inoculación traumática por
algunos tipos de hierbas o arbustos que puedan contener la
micobacteria. Sin
embargo, la transmisión en el hombre también puede venir dada por
acontecimientos naturales, como las inundaciones, ya que la
Mycobacterium ulcerans
puede crecer en aguas estancadas, así como también debido a
modificaciones del suelo como consecuencia de la acción del hombre
entre ellas las construcciones. Otra
posible forma de infección podría ser a partir de las picaduras de
insectos acuáticos. Los cuales habitan en las raíces de plantas
acuáticas de regiones con climas templados y tropicales, ricos en
agua fresca. Por otra parte, se considera que la transmisión persona
a persona es un fenómeno excepcional. Su
patogenia comienza con su entrada en el tejido cutáneo, donde va
generalmente a producir un nódulo o una pápula cutánea indolora
que en ausencia de tratamiento, evoluciona hacia una ulceración
masiva de la piel y en ocasiones afecta a las extremidades. Esto
sucede como consecuencia de la producción de una toxina necrotizante
(micolactona o mycolactona) por parte del M.
ulcerans, actuando como
inmunosupresora local que tiene afinidad en los adipocitos con efecto
citotóxicos sobre ellos. La necrosis resultante proporciona un medio
que, al favorecer la proliferación del microorganismo causante,
determina la aceleración de la propia necrosis. Durante
esta fase necrótica, la respuesta celular del huésped es muy débil
o inexistente en un determinado momento y por un mecanismo
desconocido, la toxina puede resultar neutralizada, o bien las
bacterias dejan de proliferar y de producirla.
La
curación parece iniciarse cuando el huésped desarrolla una
inmunidad celular contra los componentes del Mycobacterium
ulcerans esto se evidencia por
la formación de unos granulomas los cuales destruyen a las
micobacterias y la enfermedad remite dejando cicatrices.
En
caso de que se observe un elevado número de bacterias extracelulares
y de una escasa respuesta inflamatoria en las lesiones causadas por
Mycobacterium ulcerans sugiere
que los individuos que desarrollan la enfermedad fagocitan de forma
ineficaz o no consiguen fagocitar las micobacterias antes de ser
destruidas, procesadas y presentadas a los linfocitos T.
La
inmunidad celular y del equilibrio en los distintos patrones de
secreción de citocinas en las respuestas Th1 y Th2 juegan un papel
fundamental en el individuo que presenta la úlcera, ya que un cambio
del patrón de citocinas de una respuesta Th1 (IFN-γ, IL-2) a una
respuesta Th2 (IL-4, IL-5, IL-12) y un predominio innato o adquirido
de respuesta Th2 frente a una respuesta Th1 defectuosa podría
facilitar el desarrollo de la enfermedad o condicionar sus
características clínicas y evolutivas. También
se relaciona con mutaciones en los genes que codifican la síntesis
de citocinas Th1 (como por ejemplo la IL-12) y/o sus receptores que
son el receptor del interferón gamma [IFN-γ R] o receptor de la
interleucina 12 [IL-12R]. Las
mutaciones homocigotas de dichos genes dan lugar a un déficit
parcial o completo de la vía de citocinas Th1, el cual es un factor
activador importante de los macrófagos, por consiguiente los déficit
completos dan lugar a infecciones precoces y letales con una escasa
respuesta granulomatosa, mientras que los defectos parciales pueden
manifestarse en edades más avanzadas, presentando un mejor
pronóstico y dando lugar a lesiones con la formación de
granulomas.(19) Los
pacientes con úlcera de Buruli desarrollan una respuesta humoral
inmune frente a M. ulcerans,
evidenciada por la producción de anticuerpos específicos (con un
aproximado de un 70%). Sin embargo, presentan una intensa anergia
cutánea sistémica de células T frente a antígenos de dicha
micobacteria. Desconocemos los mecanismos patogénicos implicados en
dicha anergia, aunque se ha postulado tanto la participación de la
Mycolactona
como de alteraciones inmunológicas y modificaciones en los patrones
de producción de citocinas. Induciendo
inmunosupresión local en el paciente, inhibiendo las respuestas
inflamatorias, detención en el ciclo celular G0/G1, las citocinas
proinflamatorias como TNF-
e IFN-
las cuales se ha demostrado que se expresan durante todas las
diversas etapas de la infección y por último tiene la capacidad de
producir apoptosis de las células.(1) Sin
embargo la
Mycolactona
puede causar la úlcera en sitios lejos de los sitios de infección,
induciendo una respuesta leve inflamatoria, daño de las fibras
musculares, producción de una fibrosis, en donde se ve evidenciado
un aumento de los mastocitos los cuales producen una respuesta
proinflamatoria con una síntesis de citocinas y factores de
crecimiento junto al reclutamiento de los leucocitos en el músculo
esquelético. Cuando
la respuesta inflamatoria ocurre en los sitios lejos de la infección
se crea la proliferación celular y la migración del músculo
esquelético y desgranulación de mastocitos, estos hechos pueden
ocurrir en cualquier momento de las fases en que se presenta la
fibrosis. Normalmente los subconjuntos de los leucocitos neutralizan
la fagocitosis bacteriana y actúan como presentadoras de antígeno a
las células T citotóxicas y helper, pero se a ha demostrado que la
presencia crónica de neutrófilos y macrófagos en el sitio de
lesión explica que no se pudo eliminar el agente causante por lo que
se ve evidenciado la capacidad de
Mycobacterium ulcerans
para inhibir los pasos de resolución de la inflamación.
Una
úlcera se va a desarrollar en tres etapas, inicialmente se empieza
con la presencia de un nódulo, pápula o edéma en el sitio de
lesión correspondiendo a la primera fase de la infección, el cual
es indoloro y pruriginoso. En
ausencia de un tratamiento la lesión va a evolucionando creando una
úlcera en la piel sin bordes definidos la cual corresponde a la
segunda etapa de la lesión. Posteriormente
se empieza la tercera fase que corresponde a una curación espontánea
y formación del granuloma; en algunos casos se puede presentar una
cuarta fase que es la presencia de una fibrosis. En
algunos casos estas lesiones pueden causar complicaciones
incapacitantes como
deformidades por contractura, amputación de miembros y pérdida de
órganos (ojo, mama, genitales). Se han descrito algunos casos de
fallecimiento debido a septicemia, tétanos o hemorragia. También se
ha notificado un número creciente de infecciones óseas que
complican el tratamiento de estos casos. Pueden deberse a la
propagación directa desde la lesión cutánea suprayacente, o
avalar la hipótesis de la diseminación hematógena. (19) Mycobacterium
ulcerans tiene la capacidad de
formar biofilms, definidos como comunidades de microorganismos que
crecen en una matriz de exopolisacáridos y adheridos a una
superficie inerte o un tejido vivo, produciendo a la bacteria una
resistencia contra factores ambientales contrarios y los
antimicrobianos.(6,20)
En
la actualidad no se conoce
exhaustivamente la tasa de morbilidad a nivel mundial y de los
países que presentan la úlcera de Buruli. Por lo general, la
enfermedad afecta sobre todo a los habitantes pobres de zonas rurales
alejadas, en las que el acceso a la atención de salud es limitado
especialmente a los niños menores de 15 años. Ningún grupo racial
o socioeconómico está a salvo.(21)
Recapitulando
sobre la patogénesis toxigénica de la úlcera de Buruli, se han
descrito una familia de moléculas solubles e hidrofóbicas, lípidos
macrocíclicos, estables conocidas como las Mycolactonas
que actúan como toxinas con un efecto tanto citotóxico como
inmunodepresor local, esta penetra en las células por difusión
pasiva y se acumula en el citoplasma inhibiendo así la síntesis
protéica; inducen
la apoptosis de los macrófagos e inhiben tanto la proliferación de
linfocitos T como la producción de interleucina 2 por las células T
y de TNF-α por parte de los monocitos; la anergia observada en la
úlcera de Buruli podría ser debida en parte por la acción de esta
Mycolactona.
Se han implicado, asimismo, otras enzimas, una fosfolipasa C y una
hemolisina (que induciría la producción de TNF-α) que darían
lugar a efectos tóxicos en la infección por M.
ulcerans. No ha podido
establecerse ninguna correlación entre la cantidad o tipo de
micolactona y la intensidad o evolución y forma clínica.
Estas
observaciones parecen sugerir que el número y el tipo de
mycolactonas implicadas podrían influir en el desarrollo de patrones
clínicos distintos. Sin embargo, la participación de otros factores
acompañantes (biológicos, económicos, culturales, disponibilidad
de recursos sanitarios) puede, asimismo, ser fundamental. A
pesar de conocer la acción de la micolactona en el organismo se
desconocen los mecanismos intrínsecos de la respuesta inmunológica
en la infección por M. ulcerans.
La existencia de una anergia de células T frente a M.
ulcerans parece representar un
fenómeno importante en
el desarrollo de la úlcera de Buruli. Se ha demostrado
que las células mononucleares
de la sangre periférica de
los pacientes con enfermedad activa o tras la extirpación
quirúrgica presentan tanto una
proliferación linfoide como
una producción de IFN-γ reducida en respuesta
frente a las micobacterias. Esta
anergia parece
ser adquirida y puede persistir durante años después
del tratamiento y resolución de
la lesión. El
IFN-γ es un factor importante en la activación macrofágica y
desempeña un papel fundamental en la protección frente a las
micobacterias. Una baja producción de IFN-γ en pacientes con
infección por M. ulcerans podría
ser, en parte, la causa de la persistencia de las micobacterias
extracelulares, la evolución indolente de la enfermedad y la falta
de respuesta al tratamiento antibacteriológico convencional. Se
ha sugerido que una elevada producción de IL-10 por las células T
con un patrón Th2 podría estar implicada en la baja respuesta de
IFN-γ específica frente a M.
ulcerans. Se han demostrado
perfiles distintos de expresión de citocinas en las distintas fases
de la enfermedad: la presencia de nódulos se asociaría con una
producción mayor de IFN-γ y menor de IL-10, mientras que en las
fases ulceradas se objetivaría unas concentraciones menores de IFN-γ
y una producción superior de IL-10. Una
producción inadecuada de citocinas puede desequilibrar el sistema
inmunitario de los pacientes que desarrollan una infección por M.
ulcerans. En el momento del
desarrollo de la enfermedad se ha descrito un cambio del patrón de
citocinas de una respuesta Th1 a una respuesta Th2, que facilita la
producción de anticuerpos específicos, por lo que han sugerido que
las respuestas Th1 pueden prevenir el desarrollo de la enfermedad.
Un
mejor conocimiento de los mecanismos patogénicos implicados en el
desarrollo de estas lesiones puede permitir en un futuro plantear
posibles intervenciones terapéuticas mediante citocinas que
estimulen una respuesta Th1. Un patrón de producción de citocinas
predominantemente Th2 da lugar a una inhibición de la actividad
macrofágica, y lleva a una disminución de la resistencia contra
diferentes microorganismos. Una respuesta inmunológica de tipo Th2,
que aumentaría la susceptibilidad de los individuos al desarrollo de
úlcera de Buruli, tras entrar en contacto con el agente causal. El
TNF-α podría ser otro factor adicional importante en la activación
de los macrófagos, que fagocitan y destruyen las micobacterias. El
TNF-α es un factor fundamental en la protección frente a
infecciones por micobacterias, esto se evidencio ya que los ratones
deficitarios en TNF-α son altamente susceptibles a la infección, y
no consiguen desarrollar una respuesta granulomatosa.(21) Sin
embargo, no se ha podido demostrar de forma definitiva su papel en el
desarrollo de las lesiones cutáneas, en la trombosis subyacente y en
la necrosis de los adipocitos.(18)
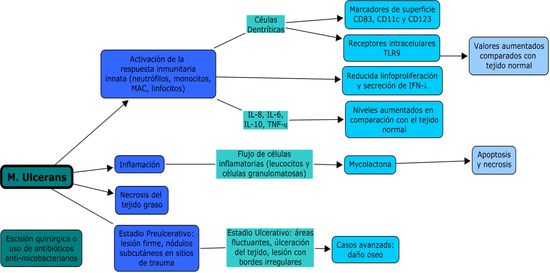
En
la figura 3 se presentan resumidos las fases de la infección por M.
ulcerans y la acción de su
toxina. 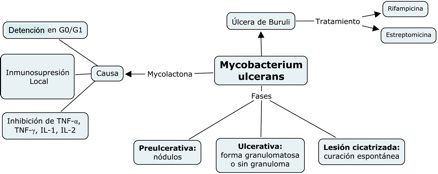
Figura
3.- Fases patogénicas de la
infección por M. ulcerans
El porcentaje de macrófagos y de linfocitos
CD4/CD8 encontrados en el sitio de lesión, junto con su expresión de de IFN-γ,
IL-10, TNF-α Y TGF-β, fue relacionada
con la respuesta inflamatoria
evaluada por histopatología. Todos los casos de úlcera de Buruli
mostraron una necrosis extensiva e inflamación crónica. La característica más
importante fue la presencia o ausencia de granulomas coexistentes junto con un
balance mixto de citocinas pro-inflamatorias/anti-inflamatorias.
Cuando los granulomas estaban
presentes se veía una significativamente alta expresión de IFN-γ; mientras que
en lesiones ulcerativas sin granulomas había un aumento en la expresión de
IL-10 y un mayor contaje bacilar.(21)
Estas características se correlacionan con la
cronicidad de la lesión, las lesiones de mayor duración mostraron granulomas,
Así que, los granulomas estaban ausentes en las lesiones ulcerativas
relativamente tempranas, las cuales contenían
muchos bacilos y poco IFN-γ, esto sugiere que durante esta etapa de la
enfermedad una fuerte supresión de la respuesta inmune celular facilita
proliferación bacilar.(19)
La figura 4 esquematiza la
respuesta inmune en la formación de la úlcera de Buruli. Figura
4.- Efectos de la infección
por M. ulcerans en el huésped
Por
todo esto se considera que la
reacción inmunitaria del huésped desempeña un papel importante actuando como
factor determinante de la extensión de la lesión y es interesante señalar el
hecho de que en el caso de muchas lesiones, se piensa que estas curan
espontáneamente. La ulterior acumulación (y/o
inducción) de la toxina puede ser lo bastante importante como para provocar la
lisis del macrófago huésped y paralizar las funciones celulares de los
linfocitos y los macrófagos infiltrantes. A su vez, esta inmunosupresión local
podría contribuir a retrasar la reacción inmunitaria sistémica precoz al
antígeno bacteriano, lo que explicaría el hecho de que, a menudo, los pacientes
con lesiones evolutivas no reaccionen a la administración intradérmica de un
antígeno derivado del M. ulcerans (conocida como la prueba de la
burulina).
Más adelante, en la fase de
curación caracterizada por la aparición de granulomas, la conversión de la
intradermorreacción a la burulina a la positividad indica que se desarrolla una
respuesta celular específica. La reacción de la inmunidad celular desempeña un
papel importante en la curación. Cabe destacar que no se observó reacción en las fases iniciales
de la infección por el M. ulcerans, mientras que se obtuvieron
respuestas positivas en los pacientes con lesiones en vías de curación.
Las investigaciones sobre las
reacciones mediadas por anticuerpos frente a las micobacterias no han dado
frutos debido al solapamiento antigénico en este grupo de microorganismos y a
su ubicuidad en la naturaleza. Sin embargo, la patogénesis de la infección por
el M. ulcerans suscita una importante cuestión: si la Mycolactona inhibe el desarrollo de una
reacción inmunitaria eficaz, ¿cómo logra el huésped superar esta situación
para, a la larga, curarse? Una explicación sería que la toxina es neutralizada
por anticuerpos que van produciéndose lentamente. Al poseer una estructura de
macrólido, es improbable que la Mycolactona
induzca la síntesis de anticuerpos.(19)
Úlceras por microorganismos diferentes a
micobacterias.
Algunos
microorganismos son productores de biopelículas o biofilms, figuran entre
ellos Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus,
entre otros.
El ejemplo clásico
y adaptable a la patología ulcerativa crónica es la Pseudomonas aeruginosa un
bacilo gramnegativo no fermentador. Es una bacteria intrahospitalaria oportunista, produce infecciones
cuando el huésped tiene alteradas las defensas, principalmente en pacientes con
quemaduras extensas, leucémicos, transplantados.(22)
Su hábitat son las
fuentes de agua ambientales, piel, sistema respiratorio superior, colon,
respiradores hospitalarios y humidificadores.(23)
Fue aislada en un 18.6% de las veces de úlceras de
pacientes VIH-positivos, procedentes del Servicio de Dermatología del Hospital
Vargas de Caracas.(24)
Entre sus determinantes de
patogenicidad se encuentra la presencia de un pili, y dos toxinas: la
endotoxina, responsable de la fiebre y del shock séptico asociado a la sepsis.23
La exotoxina A, la más importante, que inhibe la síntesis proteica en las células
hepáticas, corazón, riñón, pulmón y bazo. Inhibe la captación de aminoácidos a
nivel celular.
Dentro de la gran variedad de patologías que produce
pueden mencionarse: endocarditis de válvula nativa y protésicas, neumonía
intrahospitalaria, meningitis, septicemia en neutropénicos y quemados.(23)
Las
úlceras crónicas en las piernas son colonizadas por bacterias saprofiticas,
estas no influyen en la curación de la úlcera; otros microorganismos no se
comportan de la misma manera con respecto a la formación de tejido sano.(25,26)
P.
aeruginosa ocasiona crecimiento de la herida, a pesar del tratamiento
antimicrobiano. Se demostró que
el deterioro de la úlcera fue causada por la invasión al tejido circundante,
causando el crecimiento de la ulcera y la formación de anticuerpos. La
exotoxina A ejerce efectos patogénicos en el retraso de la curación de las
heridas y restauración de los procesos normales de curación con anticuerpos de
exotoxinas A. Sin embargo, el estudio señaló que la cantidad de exotoxina no
está relacionada con el tamaño de la úlcera.(25)
LLLLos biofilms son
grupos de bacterias mantenidas en sustancia
polimérica extracelular asociados
a una superficie responden parcialmente los antimicrobianos. Sin embargo, al
ubicarse en espacio de la herida causan cronicidad y perpetúan la inflamación
asociada.
Las hipótesis que
explican la permanencia de los biofilms plantean que el huésped desarrolla una
respuesta hiperinflamatoria incapaz de superar la barrera, nutre las bacterias
y las perpetua, convirtiendo la herida en crónica. El biofilm interactúa con el huésped en un enlace estable de nutrición
en forma de relación parasitaria.(6,27)
El biofilm
secuestra el sistema inmunológico del hospedador siendo capaces de estimular
las células, del hospedador, para
producir citocinas en exceso dañando los tejidos. Por esta razón estas
infecciones corresponden a la ruptura de los conceptos de universalidad (inmensa cantidad de respuestas inmunes pueden generarse en contra de
cualquier reto potencial), tolerancia (barreras mayores que previenen o limitan
la respueta a lo propio y a los no
patógenos), apropriateness (respuestas
que tienen solo como blanco a los patógenos. La magnitud de la respuesta
es apropiada con el nivel de amenaza, haciendo un esfuerzo por no causar daño
en el huésped), por los cuales se
rige el sistema inmunológico.
La detección de
biofilm en el organismo estimula la secreción de NF-kB lo que se reconoce por
el huésped como señal de alerta por invasión patógena. El sistema inmunológico
responde ante esta señal con la migración de neutrófilos al área de la herida,
promueven la liberación de citocinas proinflamatorias GM-CSF, IL-1, TNF-a, IFN-g e IL-8.27 Lo anterior se expresa en
forma resumida en la figura número 5.
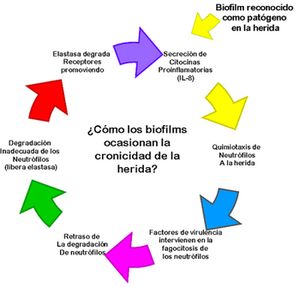 Figura 5.- Secuencia de eventos donde los biofilms ocasionan la
cronicidad de la herida Figura 5.- Secuencia de eventos donde los biofilms ocasionan la
cronicidad de la herida
La literatura indica un exceso de neutrófilos acumulados en el sitio de
las heridas crónicas y otras infecciones crónicas. La IL-8 en la herida
promueve la quimiotaxis de los neutrófilos, en un sistema inmunológico sano,
estos inician la apoptosis luego de engullir al patógeno, expresando marcadores
de superficie que son reconocidos por los macrófagos. Los productos bacterianos
como los lipopolisacáridos interfieren en la expresión de estas moléculas de
superficie evitando así el inicio de la apoptosis de los neutrófilos y de la
fagocitosis por parte de los macrófagos.
La degradación posterior ocasiona la liberación de proteinasas y
elastasa que interfiere en la cicatrización degradando el tejido de la matriz.
La elastasa degrada los receptores del neutrófilo CXCR1 cuyos fragmentos
estimulan receptores TLR2 a producir más citocinas inflamatorias, ocasionando
la cronicidad de la herida.
Las
heridas crónicas infectadas deben ser desbridadas quirúrgica o enzimáticamente
para eliminar el tejido necrosado y la película bacteriana. Los antisépticos
tienen una limitada eficacia y la resistencia a antibióticos es 1000 veces
mayor para las bacterias de la película que en bacterias de fase planctónicas.
Las bacterias se comunican mediante un sistema de señales químicas (señales quorum sensing) para iniciar la síntesis
de biofilm y de factores de virulencia (6). La interferencia de
estas señales ésta siendo considerada en los tratamientos de las infecciones
crónicas. En previos estudios básicos se ha determinado que Lactobacillus
plantarum y sus productos inhiben tanto in
vivo como in vitro la capacidad
patogénica de P. aeruginosa. En
estudios clínicos preliminares demostramos que L. plantarum es efectivo
en el tratamiento de úlceras y quemaduras infectadas.(28)
|