The
ASM thickening has been studied in many animals and human models, wherein ASMC
cultures have provided some ideas about pathways underlying the origin of
hyperplasia and hypertrophy. Once ASMC populations were characterized in vivo and in vitro, heterogeneous subgroups with distinctive phenotypes were
identified. A wide range of functions depend on culture conditions(60). Manipulating such environments
allowed comprehension of the rules for phenotypic transition(61, 81, 82). Hence, ASMC could be sorted
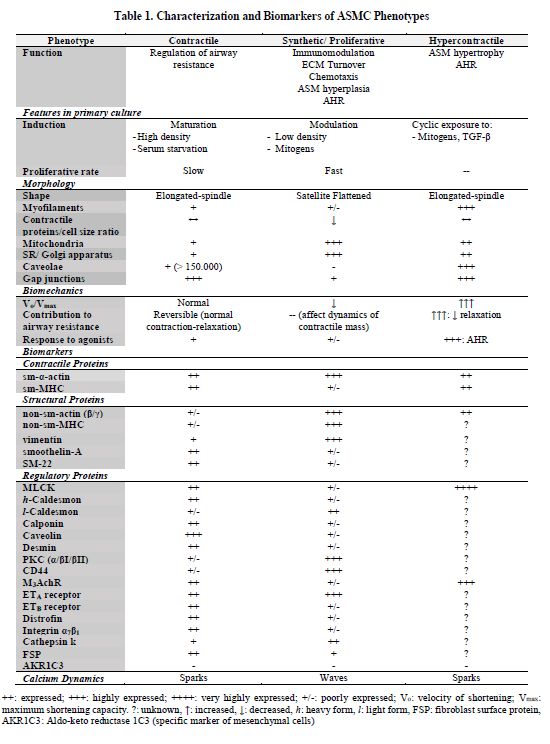
into three categories: 1) contractile (c-ASMC), 2) synthetic/proliferative
(s/p-ASMC), and 3) hypercontractile (h-ASMC) (see Table 1). Also, a few switching routes have been described, where modulation means a shift from
contractile to synthetic/proliferative, and maturation
is the inverse transition. Turning into hypercontractile is also possible, and some
authors have speculated about its irreversibility; however, in vitro ASMCs can tolerate cyclic
phenotypic adjustments. An important aspect is that modulation and maturation exemplify
an adaptation model to tissue microenvironment fluctuations, events that could take
place in vivo and drive critical
phases of airway remodeling. Accordingly, transition from native c-ASMC to
s/p-ASMC would be the initial step, then, replication of s/p-ASMC would warrant
smooth muscle hyperplasia, and finally aberrant differentiation from either
s/p-ASMC or c-ASMC to h-ASMC would cause muscle hypertrophy. How these
phenotypical modifications fit in the natural history of airway diseases is a
matter of debate.
Phenotypic Markers
Smooth
muscle has typical features in primary cultures (see Table 1, Fig. 3). A long cellular body, central nucleus, few
granulations, and cytoplasmic inclusions (3A),
a confluent monolayer with hill and valley aspect (3C), and shrunk reaction to contractile agonists define smooth
muscle cells(83). However, each
ASMC subpopulation has specific characteristics. For example, the synthetic/proliferative
phenotype has satellite flattened shape with multiple extensions (3B), a high number of organelles for
protein and lipid synthesis, abundant mitochondria, a higher proliferative
response, decreased contractile proteins, shutdown of responses to contractile
agonists, and secretion of growth factors, collagen, cytokines, bradykinin, and
eotaxin(81,84). Furthermore,
modulated ASMCs show increased protein expression of fetal and non-muscle
isoforms. Synthetic and proliferative functions do not correspond to different traits.
Indeed, the cell distribution with synthetic activities could vary between 20
to 60%, and almost half of replicating ASMCs produce cytokines. Also, secretion
can be done by non-replicating cells(81).
On the other hand, the contractile phenotype is associated with a decreased number
of synthetic organelles, a stronger response to contractile agonists, increased
expression of contractile and structural proteins, and an increased M3/M2
muscarinic receptor expression rate(84-86).
Other markers including Ca2+ profiles(87), miRNA expression(88),
and transcription factors expression(89)
have been useful for phenotype distinction.
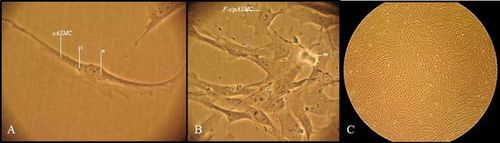
Figure
3. Primary cultured of Rat ASMCs. Cells were obtained by enzymatic
digestion of rat trachea and cultured in supplemented medium as previously described(122) . (A)
Contractile phenotype, (ci) cytoplasmic inclusions, (n) cell nucleus. (B) When the
cell population underwent to growth some cells adopted a myofibroblast-like
morphology (F-s/pASMC), (m) mitosis. (C) Cell confluence of 80-90%, cell
population readopted contractile morphology with a hill and valley array. Magnification
400X A, B, 100X C.
Evidence of in vivo Plasticity
Several
findings support in vivo occurrence
of ASMC plasticity, especially in asthma. Plasticity is a universal property of
primary ASMC cultures derived from both healthy and diseased humans, and healthy
and sensitized animals. Immunohistochemistry to identify contractile proteins
is highly variable as well, which can reflect a broad heterogeneity of myocytes
in the normal airway that is maintained in cell culture, as demonstrated by a divergent
proliferative capacity(90). If
functional features are compared, healthy or control vs asthmatic or sensitized
groups, significant differences can be found. ASMCs from asthmatics or
sensitized animals show more proliferative and synthetic capabilities than
their physiologic counterparts, findings that are preserved despite tissue
dissolution follow by cell culture(62, 66, 89,
91-93). Abnormal ASMCs could not only resemble s/p-ASMCs or arise
from c-ASMCs, but also have distinctive features such as: abnormal protein
synthesis(94), expression of
odd transcription factors isoforms with a lack of response to glucocorticoids(91), increased mitochondrial biogenesis
and activity(93), abnormal
calcium dynamics(92), increased
CysLTR-1 leukotriene receptor expression(55),
increased activity of promitogenic pathways(95),
and declined of antiproliferative pathways(66).
In consequence, an increased ASM mass may be explained by intrinsic alterations in pathological ASMCs that facilitate their
proliferative and secretory activities. Asthmatic ASM produces more
proinflammatory, proangiogenic, and proremodeling factors, including eotaxin,
VEGF, and connective tissue growth factor (CTGF), and fewer antimitogenic
factors, such as E2- type prostaglandin (PGE2)(59). These would reflect deeper differences
in cell populations that constitute the ASM under pathological settings, and
they would likely originate from comparable modulation and maturation events on
native ASMCs.
Many
questions arise from the alterations observed on asthmatic or sensitized cultured
ASMCs. Based on the plasticity phenomena, any transformation of in vivo ASMC phenotype which persistence
depends on tissue microenvironment should not be seen in vitro because the phenotype will adjust to culture conditions.
Although, cited studies do not precise whether those pathologic features are
irreversible along culture passages, persistence of functional abnormalities after
tissue fragmentation and culturing suggests that these cells underwent through
a dysfunctional route of phenotypic modulation, which could be at least
partially irreversible. In view of that, epigenetic mechanisms could be a
suitable explanation to such phenotypic switch. For example, eotaxin hypersecretion
by ASMCs has been related to histone H4 lysine 5 and lysine 12 acetylation at
the eotaxin promoter induced by TNF-α(96).
Other synthetic activities, such as VEGF hypersecretion, were due to a loss of
a repression complex, in which a differential histone H3 lysine 9 methylation
modulating Sp1 and RNA polymerase II binding to the VEGF promoter was
implicated(97). Binding of
serum response factor (SRF), a transcription factor that controls phenotypic
stability, to DNA is associated with post-transcriptional histone modifications
including di-methylation of lysine residues 4 and 79 on histone H3, acetylation
of lysine 9 on histone 3 and acetylation of histone H4. Histone deacetylases
(HDACs) have also been implicated in regulating smooth muscle replication because
the HDAC inhibitor TSA can prevent cell proliferation(88). In in vivo, the valproic acid (HDAC inhibitor) did not affect
inflammation induced by OVA challenges, but notably reduced the airway thickening
including the ASM with blunting of AHR(98).
In addition to HDAC modifications, DNA methylation can generate a specific long-term
signature. For example, expression of
IL-13 in the airways ensued significant changes in methylation of 177 genes,
most of which were associated with a Th2 signature over resident cells(99). Using methylated
DNAimmunoprecipitation-next generation sequencing (MeDIP-seq), it was determined
that airway remodeling and AHR in house- dust- or mite-sensitized rats are
related to specific methylation patterns at several TGF-β signaling-related
genes(100), explaining the
longevity of abnormal pro-fibrotic responses of local cells during inflammation
and phenotypic persistence after tissue extraction. Unfortunately, there is
currently no direct evidence of epigenetic regulation of ASMC proliferation.
Moreover, whether or not DNA methylation or histone acetylation can influence
phenotypic switching have to be determined as well. The contribution of miRNAs
will be discussed in following sections.
Potential Sources of ASMCs
The
in vivo source of ASMCs under
pathological conditions is unclear. ASM may originate from increased
proliferation or prolong survival of preexisting smooth muscle with
proliferative and/or contractile phenotype; however they could also arise from
other cell lines that could migrate into the bundles and then differentiate
into ASMCs (see Fig.4). Remarkably,
other airway cells may undergo to phenotypic modulation that is characterized
by α-sm-actin expression and development of organelles for synthetic functions.
Accordingly, mesenchyme such as fibroblasts may generate myofibroblasts, whose
classical phenotypic markers are indistinguishable from s/p-ASMCs(101). This fact allowed researchers to
postulate a spectrum of mesenchymal plasticity (fibroblasts ↔ myofibroblasts ↔
ASMCs)(102). However, it does
not undermine experimental findings obtained with in vitro systems, as a high proportion (~60%) of primary airway
mesenchymal cultures truly correspond to primary smooth muscle(60).
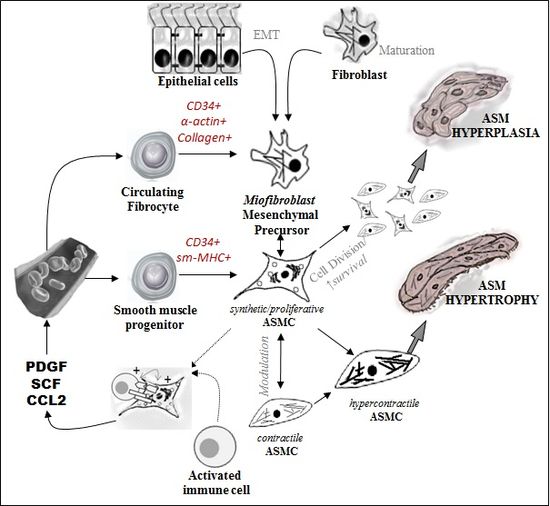
Figure
4. Potential sources of ASMC precursors in the origin
of ASM thickening. (See the text for explanation).
Potential
progenitors also include true multipotent mesenchymal progenitors and stem
cells, either located within the airway or derived from peripheral blood. For
example, CD34+-CCR7+-Collagen 1+-sm-α-actin+
circulating fibrocytes can migrate towards ASM bundles during inflammatory
challenges, and they were unresponsive to the apoptotic effects of
glucocorticoids in culture(103).
Fibrocyte migration is directed by the ASM-derived PDGF and CCL2, and at that
point its co-locating induces proinflammatory activities in ASMCs(104, 105). A rare population of CD34+-sm-MHC+
peripheral mononuclear cells (known as smooth muscle progenitors) has been
identified by flow cytometry in OVA-sensitized mice. A similar population seems
to generate the smooth muscle in atherosclerosis; however, the study did not
precise whether stem cell homing occurred into ASM bundles(106).
The
airway epithelium can turn into mesenchymal cells through the
epithelial-mesenchymal transition (EMT) route, which has been considered as
another source of ASMCs(107), but
a linage-tracing study suggests that it may just be a consequence of culture conditions
and could not occur in vivo(108). In asthma, epithelial cells show
fragileness due to downregulation of cell adhesion molecules, which makes EMT
more likely(109). EMT is
initiated by extracellular signals, such as collagen or hyaluronic acids, and
by growth factors like TGF-β and EGF(110).
This process is modulated by bone morphogenesis proteins, and allergen
exposure, which amplifies and accelerates it(111). Hormones have also been associated, since vitamin D
attenuates TGF-β-induced expression of EMT markers(112). Epithelial and mesenchymal cells express both type-1
and type-3 muscarinic receptors (M1, M3)(113). TGF-β-induced EMT was abolished by
muscarinic receptor (mAChR) antagonists and enhanced by acetylcholinesterase (AChE)
inhibitors(114). A positive
feedback loop of autocrine and paracrine production of non-neuronal acetylcholine
(ACh) and TGF-β orchestrates EMT during chronic inflammation, being a likely
source of ASM.
Additionally, an increased number of fibrocytes was
observed in the ASM bundles from asthmatics of all severities(115). However, this study failed to show
any link with the lung function, and this location could not be considered
abnormal as fibrocytes are normal constituents of ASM bundles under
physiological conditions(116).
An increased in mesenchymal cells would be nonspecific and occur in parallel to
other cellular changes ongoing in the ASM bundles. Although, many airway cell
lines can follow similar modulation pathways as native ASMCs, we focus here on
how the behavior and responses of c-ASMC vs s/p-ASMC can explain many abnormal
structural and functional features seen on airway diseases.