Signaling Pathways Associated with
Maturation
Phenotypic
switching is regulated by growth factors, GPCR agonists, ECM molecules, and
other mediators found in the bronchoalveolar lavage (BAL) of patients with
asthma or COPD(133,134). The
contractile phenotype induction is attainable after exposure to either TGF-β,
insulin, or laminin(135-138).
Also, maturation can be supported by lacking of mitogens; in primary ASMC
cultures (see Fig.3C), high cell
confluence lead to cell cycle arrest by cell contact(82, 86). sm-specific protein expression
is enhanced by RhoA/Rho Kinase and/or PI3-K activation. RhoA/Rho kinase
promotes actin polymerization as a downstream effector of either GPCR- or
RTK-associated pathways (see Fig.5).
Subsequent phosphorylation events, RhoK→ phospholipase D (PLD) → LIMK-1→
cofilin could be responsible for G-actin polymerization into F-actin(139). A decrement in globular actin level
releases some proteins into the cytosol, like the transcriptional coactivator
megakaryocytic acute leukemia/ megakaryoblastic leukemia-1 (MAL/MKL-1), which
can be trafficked into the cell nucleus and make a macrocomplex with both myocardin
(other coactivator) and SRF, a transcription factor. As a whole, they bind gene
promoters to increase sm-specific gene expression(140, 141). TGF-β can amplify this pathway for
hypercontractile phenotype induction. Type-1 and type-2 TGF-β receptor
activation induce phosphorylation and nuclear translocation of Smad proteins
that bind to SRF, building up a macrocomplex similar to MAL/MKL-1/myocardin/SRF,
with subsequent gene expression modifications(142).
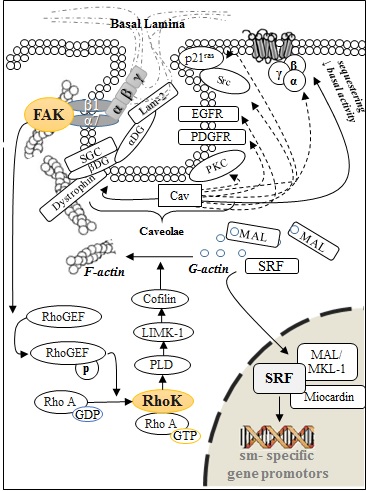
Figure 5. Signaling pathways involve in
preserving the ASM in a differentiated state.
The
Akt/PI3-Kpathway also affect sm-specific gene expression through the
transcription factor FOXO-4(143, 144).
Unphosphorylated FOXO-4 binds myocardin and inhibits its association with SRF.
Hence, myocardin is released once PI3-K phosphorylates FOXO-4. However, the reach
of this pathway goes far beyond transcriptional regulation. Signaling pathways
that converge to ribosomal regulation are needed to complete ASMC maturation,
which include effectors such as PI3-K, Akt-1, mammalian target of rapamycin (mTOR),
and p70 ribosomal S6 kinase (p70S6K). Pharmacologic inhibition of
PI3-K and mTOR are enough to prevent p70S6K activation and
sm-specific protein accumulation(143).
Moreover, activated mTOR can phosphorylate eIF4 binding protein-1 (4E-BP1), releasing
and increasing the eukaryotic initiation factor-4 (eIF4) activity(145), followed by contractile protein
accumulation (see Fig. 6B).
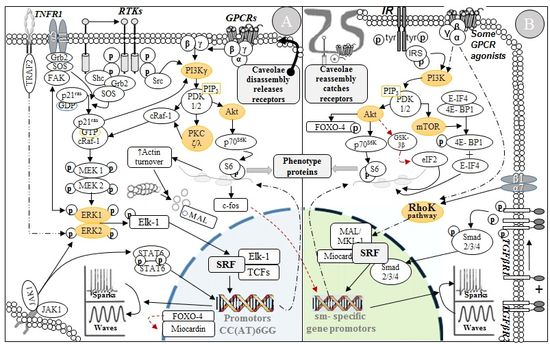
Figure
6. Signaling pathways involve in the control of ASM phenotypes. (A) Modulation, (B) Maturation.
Persistence
of the c-ASMC phenotype is highly dependent on caveolae membrane system and
cav-1 expression (see Fig. 5)(146). These flask-shaped structures are classically
considered as special compartments for signaling regulation. Cav-1 is located
inside those microdomains, being responsible for their formation and
maintenance. Interestingly, cav-1 leads to reduced basal activity and
sequestration of several receptors and signal transducers related to
synthetic/proliferative phenotype induction, such as: PKC, PDGFR, EGFR, Src,
and p21ras(147). Proteins such
as α-subunit of G proteins, Rho family members, adenylate cyclase isoforms, 7TM
receptors, and others with binding domains to glycosyl- phosphatidylinositol
are regulated as well. Caveolae act on behalf of cell adhesion by linking the
actin cytoskeleton with the basal lamina, facilitated by laminin-2/α7β1
integrin interaction. The triggered downstream pathway activates a guanine
nucleotide exchange factor, RhoGEF, leading to Rho Kinase activation(147). For this reason, it is possible
that mitogen stimulation is not enough to ignite ASMC modulation and cell
division, thus caveolae disassembly would be a strict requirement(148).
Laminin
is a trimer that bind integrins and non-integrin receptor subtypes, including the
dystrophin-glycoprotein complex (DGC). PI3-K inhibition prevents both ASMC
maturation and accumulation of DGC proteins, β- and α-DG(149). Preferential expression of DGC in
c-ASMC is related to a tighter regulation of reactions to contractile agonists
by caveolae system. There is also evidence suggesting that DGC, through β-DG, influences
signal transduction by scaffolding properties and interactions with cav-1(150). Additionally, basal activity and
low grade stimulation of some RTKs, such as the insulin receptor (PI3-K
pathway) and GPCRs (Rho Kinase pathway), could have a role in contractile
phenotype conservation, especially in confluent cells. These observations
acquire more relevance considering that the ASM mass is likely composed of
c-ASMC under physiological conditions. In the disease-setting, ECM component dissolution
by MMPs affect the ASMC attachment, shutting down the Rho kinase activity. Unconstrained
ASMCs are more susceptible to paracrine influences. Thus, high levels of
cytokines and growth factors, and subsequent anomalous repair due to TGF-β, might
match with a continuous and dynamic process of phenotypical modulation
(contractile → synthetic/ proliferative → hypercontractile/ fibrotic),
explaining the ASM thickening and dysfunction.
Signaling Pathways Associated with
Modulation
Transition
to a synthetic/proliferative phenotype is enhanced by mitogens such as PDGF,
EGF, IGF, fibronectin, collagen-I and -II, bradykinin, GPCR agonists, cigarette
smoke extract, lipopolysaccharide (LPS), and reactive oxide species (ROS)(151, 152). Modulation is quickly reached
when in vitro ASMCs are not confluent
under mitogenic influences (see Fig. 3B),
especially fetal bovine serum (FBS) and fetal calf serum (FCS)(86, 122). Associated pathways converge
to increase c-fos (coactivator) expression, which paradoxically needs a prior
SRF activation in order to alter gene expression(144). Nevertheless, this apparent duality could be due to a
wide variety of transcriptional coactivators that assertively induce selective
gene transcription. Contrasting the SRF-myocardin complex effect, SRF cooperativity
with ternary complex factors (TCF) such as Elk-1 affects gene promotors with a
CArG (CC(AT)6GG) sequence, inducing cell proliferation instead of
maturation. In this way, c-fos upregulation is key to halt contractile gene
expression(153). Furthermore,
this coactivator Elk-1 is phosphorylated and activated by the extracellular
signal- regulated kinase (ERK)-1 and -2(154). These kinases along with p38, c-Jun
N- terminal kinase (JNK), janus kinases (JAKs), and
transcriptional factors, like NFκB and AP-1, could participate in signal
delivery to increase synthetic and proliferative activities(154). Protein synthesis associated with
modulation is also favored following S6 ribosomal subunit phosphorylation(138). Increased cytoskeleton metabolism with
actin polymerization blockade generates G-actin accumulation that avoids
nuclear translocation of MAL/MKL-1. Also, it has been described that in vitro both maturation and modulation
are reversible.
ASMC
modulation is improved by Th2 cytokines. A mix with TGF-β and leukotriene D4, triggers
the expression of 29 transcription factors(155).
IL-13 is relevant to control the expression of aroung 300 locus. Its receptor,
the IL-13Rα1/IL-4Rα complex, mediates the phosphorylation of STAT-6, triggering
MAPKs for phenotypic modulation(156).
IL-13 can also affect calcium dynamics by upregulation of sarcolipin, which is
a transmembrane protein placed at the sarcoplasmic reticulum (SR) that inhibits
the sarco/endoplasmic reticulum Ca2+- ATPase (SERCA) activity(157). Expression of calcium regulatory
proteins changes with modulation, therefore, a decrease in voltage-dependent
calcium channels, ryanodine receptors, and SERCA2 levels translate into a calcium
dynamics dominated by wave-like propagations. This kind of flow contributes to
MAPK pathway activation. Ca2+ waves also affect the conformational
stability of cis elements in 5untranslated regions (UTRs) of mRNAs and
interactions between translational components, regulating protein synthesis(158). IL-13 signaling is under control
of the type-1 suppressor of cytokine signaling (SOCS-1), a protein with
chaperone properties. SOCS1 expression is decreased in asthmatic ASMCs and its
inactivation raises synthetic activities when exposure to Th2 cytokines(159). In summary, diverse signaling pathways
are responsible for driving phenotypic modulation of ASMCs (see Fig. 6A).
Muscarinic Activation leads to
ASMC modulation
ASM
thickening is attainable by persistent muscarinic stimulation(127). Both pathways, Gi/0
coupled M2 and Gq coupled M3, could generate
the activation of MAPK, Rho- kinase, and PI3-K signaling(117). Moreover, shifting from a synthetic-proliferative
to a contractile phenotype is accompanied by a decrease in M2 and a parallel
increase in M3 expression(160).
Those observations inquire whether or not cholinergic stimulation may affect phenotypic
switching. Accordingly, long-term incubation of rabbit ASMCs with ACh or carbachol
(CCh) induced a switch towards s/p-ASMC(161).
Prolonged treatment of bovine ASM strips with the methacholine also diminished
contractile protein expression(162).
Transition to a synthetic-proliferative phenotype is characterized by M3 downregulation
and blunted contractile responsiveness to cholinergic stimulation(161). In cited studies, signaling
pathways were not evaluated, but considering that cholinergic-induced mitogenesis
is related to MAPK activation, it is possible that muscarinic activation allows
the nuclear translocation of Elk-1, affecting gene expression linked to the phenotype
transition.
Role of non-coding RNAs on
Phenotypic Stability
The
miRNAs are small noncoding RNAs that have an outstanding participation in gene
expression regulation. It makes them excellent candidates to control cell
plasticity. Multiple mechanisms are involved in miRNA synthesis and gene
regulation, as it was previously described(163).
Shortly, mature miRNA is part of the active RNA- induced silencing complex
(RISC) that mediates miRNA/mRNA interaction in a specific fashion. This
interaction mostly occurs in the 3UTR by partial complementarity, thus, miRNAs
inhibit elongation during translation, or destabilize mRNA promoting its
degradation. In smooth muscle biology, multiple miRNAs regulate cell
differentiation and proliferation, under physiological and pathological
conditions, especially in vascular smooth muscle, although little is known about
ASM (see Table 2)(164).
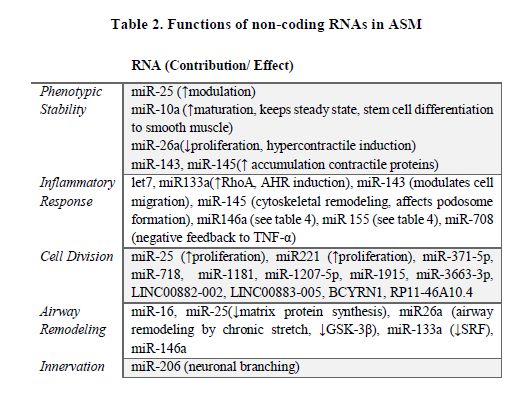
Around
11 miRNAs are upregulated in cytokine-exposed ASMCs. Particularly, miR-25 is
significantly modulated after prolonged OVA-challenge(165). Some cytokines through the
↑miR-25/↓KLF-4 system promote ASMC modulation(166). The transcription factor kruppel-like factor-4 (KLF-4)
represses sm-specific gene expression by recruiting histone H4 deacetylase
activity to smooth muscle cell genes, thereby blocking SRF association with
methylated histones and CArG box chromatin. Next-generation sequencing
identified miR-10a as the most abundant miRNA expressed in primary human ASMCs,
accounting for more than 20% of all small RNAs. miR-10a directly suppresses
PI3KCA expression and its overexpression reduces ASMC proliferation(167). TNF-α-induced expression of miR-708
in asthmatic ASMCs is greater than in non-asthmatic. miR-708 decreased JNK,
MAPK and Akt phosphorylation and increased MAPK phosphatase-1 (MKP-1) and phosphatase
and tension homolog (PTEN)expression. It constitutes a negative feedback for TNF-α
signaling downregulation(168).
miR-133a levels were decreased in human ASMCs, along with upregulated RhoA
expression during AHR. Those findings were replicated after treatment with
IL-13(169). Sonic hedgehog
signaling blocks miR-206 expression to increase the release of BDNF by ASMCs,
coordinating branch innervation(52).
A recent study explored the RNA expression
profile in cultured ASM(170). Remarkably,
over 200 miRNAs were detected including: miR-371-5p, miR-718, miR-1181,
miR-1207-5p, miR-1915, and miR-3663-3p. These miRNAs had been previously related
to aberrant proliferation in other cells. Paradoxically, predicted targets cut
down gene expression of proteins that are known for remodeling promotion. They
also detected a specific long non-coding RNA (lncRNA) profile. lncRNAs have
recently emerged as epigenetic tools for gene expression regulation. They can
regulate miRNAs as target site decoys, can also directly bind to transcription
factors and participate in assembly of chromatin-modifying complexes as
structural components and recruiters of genomic targets(171). Stimulated human ASMCs expressed
29 lncRNAs, and some of them were previously identified as cell proliferation regulators.
Relevantly, an increase in LINC00882-002 and LINC00883-005, and a decrease in
BCYRN1 and RP11-46A10.4, could explain why despite of specific miRNAs, a target
mRNA transcript is still translated. These lncRNAs could act as sponges for
the miRNAs-1207, -150, -940, and -371, blocking the RISC association with the translational
machinery. The analysis is more complex if it is considered that a variable expression
is seen in each cell cycle phase. In summary, phenotypic switching encompasses
responses exquisitely coordinated by multiple signaling pathways, orchestrating
gene expression not only at a promotor level, but also involving specific
changes in the RNA metabolism.