Microbiología
Apoptosis y Helicobacter pylori: un nuevo modelo en oncogénesis infecciosa
Protogenia del Cáncer Gástrico
Desde 1982, cuando Warren y Marshall descubrieron el Helicobacter pylori, automáticamente se dio a conocer el agente etiológico de la gastritis aguda y actualmente se asocia a diferentes enfermedades, principalmente gastrointestinales, como el adenocarcinoma gástrico, el cual ha causado mucha controversia.
El H. pylori ingresa al organismo por la cavidad oral (1). Este bacilo tiene mucha motilidad y actividad producto de su forma espiralada y flagelos polares, permitiéndole alcanzar su hábitat natural, la mucosa gástrica, tras lograr evadir los mecanismos defensivos del huésped.
Una vez dentro de la cavidad gástrica, se puede inferir que el pH ácido y las condiciones hostiles de este ambiente evitarían su estadía. Sin embrago, la ureasa, determinante de patogenicidad del microorganismo, le permite desdoblar la urea en amonio, rápida y permanentemente, creando a su alrededor un microambiente alcalino que le permite su supervivencia al neutralizar la acidez del medio(7).
En mayo del año 2000, Sachs y colaboradores identificaron una proteína a la que denominaron UREL (miembro de las amidoporinas) que regula la transformación de la urea del medio externo del estómago hacia el citoplasma del H. pylori mediante canales transmembrana; los cuales a medida que disminuye el pH incrementan en número, llegando alcanzar hasta trescientas veces la cantidad de urea que podía penetrar inicialmente, lo que conlleva a una suficiente producción de amonio para neutralizar el periplasma (si esta proteína se encuentra ausente disminuyen los canales para el transporte de urea y el H. pylori se hace vulnerable al pH del estómago) (17).
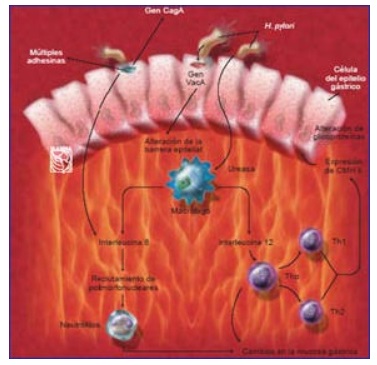
Vencida esta primera barrera el microorganismo se debe enfrentar al mucus gástrico, el cual degrada con una proteasa denominada mucinasa, especial para esa función y así puede acceder a la superficie epitelial, a la cual se adhiere fuertemente mediante las moléculas de adhesión (hemaglutininas). El H. pylori posee gran variedad de adhesinas que reconocen de forma específica los receptores de la mucosa gástrica, se unen a ellos y se promueve la liberación de toxinas directamente sobre estas células y propiciando la colonización bacteriana (7).
Entre las toxinas liberadas se encuentran: IL-1(beta), IL-8, IL-9, TNF (alfa), factor de activación plaquetaria, INF (gamma), hemolisinas, especies reactivas de oxígeno (gracias a la presencia de superóxido dismutasa), lipopolisacáridos y fosfolipasas (principales responsables del daño de la mucosa que genera la bacteria). (23)
Después de la liberación de estas citocinas, relacionadas directamente con la regulación del sistema inmune, se produce un infiltrado inflamatorio producto de la liberación de los mediadores de la inflamación, como histamina, citocinas, leucotrienos, prostaglandinas (la ciclooxigenasa y la sintasa inducible se encuentran incrementadas en las células epiteliales gástricas, durante la infección por H. pylori, lo que induce un aumento en la síntesis de prostaglandinas, leucotrienos, tromboxanos) activándose: neutrófilos, monocitos, macrófagos y leucotrienos (la ciclooxigenasa y la sintasa inducible se encuentran incrementadas en las células epiteliales gástricas, durante la infección por H. pylori) (25)
FIGURA 6: Patogenia de la infección por Helicobacter pylori
Como principales determinantes de patogenicidad, El H. pylori posee dos proteínas antigénicas:
- VacA: la polimerización de esta proteína en la superficie de las membranas lipídicas de las células del organismo, permite la formación de canales de transporte aniónico selectivo. Estos canales pueden ser endocitados y llegar luego a los endosomas activando una bomba V-ATPasa electrogénica, que permite la entrada de cloruro dentro de este compartimiento. Posteriormente el cloruro se unirá al amonio y se formará la toxina de efecto vacuolizante. Esta citotoxina disminuye la producción de ácido clorhídrico en el estómago, causando apoptosis en las células apriétales y ayudando así en la colonización. Por otra parte, se ha relacionado su difusión a través de los espacios paracelulares con la movilización de los iones níquel y hierro., sin modificar el grosor ni la permeabilidad de estos espacios. Además la proteína VacA codifica una proteína VIP-54 que induce la inhibición selectiva del MHC II, impidiendo la presentación antigénica. (ver Figura 2 y 7)
- CagA: esta proteína citotóxica, induce la producción de citoquinas proinflamatorias por el epitelio gástrico, entre las que se encuentra el MIF (factor inhibidor de la migración de macrófagos), expresado por linfocitos, macrófagos, células gástricas y monocitos (25), e induce un aumento en la expresión de su receptor, CD74. durante la infección por H. pylori, la unión de este ligando con su receptor induce la proliferación celular, e incrementa los eventos procarcinogénicos. También se le atribuye un aumento notable de la expresión de IL-8. (ver Figura 3 y 7) (26)
Secundario a todos los mecanismos patogénicos mencionados anteriormente se produce inflamación súbita del revestimiento del estómago, conocida como gastritis aguda. Si esta persiste durante un tiempo prolongado, se produce GASTRITIS CRÓNICA, en donde la inflamación afecta sólo la mucosa y puede ser asintomática. (24)
Los síntomas característicos de la enfermedad son: dolor abdominal en la parte superior que puede empeorar al comer, indigestión abdominal, pérdida del apetito, nauseas, vómitos con o sin sangre y heces oscuras.
La morfología de las lesiones se caracterizan por:
- Pérdida parcial de las glándulas gástricas, células apriétales, y las célula principales se sustituyen por células mucosecretoras. (24)
- Infiltrado inflamatorio: destacan los neutrófilos, linfocitos, células plasmáticas y a veces eosinófilos, en toda la profundidad de la lámina propia. (24)
- En las células epiteliales de superficie aparecen alteraciones, como variaciones de la dimensión forma y orientación, a veces acompañado de crecimiento y atipia nuclear. Estas alteraciones histológicas, pueden explicar un aumento de peligro de carcinoma gástrico concomitante con gastritis y atrofia gástrica (24).
Se puede dividir en dos grupos
- Gastritis crónica superficial: caracterizada por alteraciones degenerativas en las células del istmo, infiltración de linfocitos y plasmocitos preponderantemente en la porción superficial de la lámina propia, entre las foveolas gástricas; la infiltración generalmente incluye variable cantidad de neutrófilos.
- Gastritis crónica atrófica: mucosa adelgazada, con disminución de glándulas y simplificación de las glándulas remanentes; infiltración linfocitaria y plasmocitaria en todo el espesor de la lámina propia, acompañada de neutrófilos, en la mucosa fúndica puede producirse un reemplazo de las glándulas características por glándulas de tipo pilórico (metaplasia pilórica); tanto en la mucosa fúndica como en la pilórica puede haber también una metaplasia intestinal: el epitelio de las foveolas y de las glándulas está reemplazado principalmente por células caliciformes y células cilíndricas similares a las células de función absortiva del intestino (enterocitos) (22).
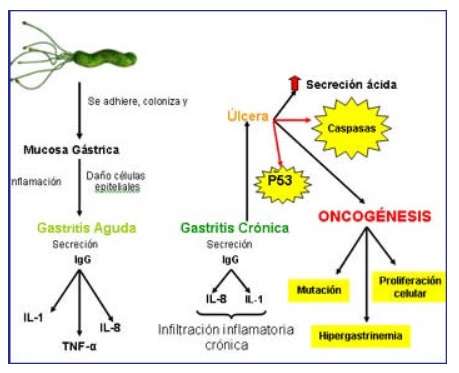
Como consecuencia de la gastritis crónica se produce una excesiva secreción de ácido clorhídrico como mecanismo de defensa por parte del organismo ante la presencia de la bacteria, que ocasiona la ruptura en el tejido normal que recubre el estómago, es decir, una pérdida focal de tejido que compromete al menos todo el espesor de la mucosa y parte de la submucosa, pudiendo extenderse a todo el espesor del órgano, denominado úlcera gástrica., exacerbándose esta patología por el aumento absoluto o relativo de ácido clorhídrico y pepsina (por aumento de la masa de las células apriétales y principales de la mucosa fúndica y mayor respuesta de las células G del antro productoras de gastrina, por aumento en su número o en su función), por encima de la proliferación epitelial, coexistiendo con hemorragias, desencadenándose episodios de isquemia focal, por apertura de anastomosis arteriovenosas de la submucosa; menor síntesis de prostaglandinas (las prostaglandinas son inhibidas por el uso de antiinflamatorios, que favorecen la aparición de úlcera péptica). Ésta puede curarse por reparación de las túnicas subyacentes a la mucosa y por regeneración atípica de la mucosa, por esta razón se asocia a la formación de maltomas y/o adenocarcinoma gástrico. (24, 22)
El aspecto característico de una úlcera activa que ha sufrido varias crisis previas es el siguiente: el fondo de la úlcera está formado, desde la superficie a la profundidad, por las siguientes capas: tejido necrótico y fibrina; polimorfonucleares; tejido granulomatoso; tejido conectivo fibroso (base de la úlcera o callo). La mucosa de los bordes presenta anaplasia de regeneración, con formación de pequeñas foveolas y una capa de epitelio aplanado que comienza a reepitelizar el fondo de la úlcera. La muscular de la mucosa y la muscular propia están interrumpidas; los cabos de la muscular propia, a ambos lados del callo, están ascendidos hacia la muscular de la mucosa. (24)
Las lesiones mencionadas anteriormente, causan un desequilibrio en los procesos apoptóticos, proliferativos, moleculares y de cualquier índole en diversas poblaciones celulares, entre las que encontramos las células gástricas, leucocitos y linfocitos; estableciéndose así lesiones precancerosas, entre las cuales encontramos las metaplasias, y la que se ha asociado en mayor grado al cáncer gástrico es el tipo Iib en el que se aprecia un mayor grado de diferenciación celular (7).
Las células epiteliales tras la adhesión bacteriana liberan IL-8 que junto con factores citotóxicos del propio germen favorecen a la activación de estos polimorfonucleares y la consiguiente liberación de proteasas y metabolitos reactivos de oxígeno (como consecuencia de la síntesis de la superóxido dismutasa), ésta situación provoca un estallido oxidativo y la activación de la cascada de las caspasas quienes escinden el ADN induciendo mutaciones en las células germinales mucosas que de no repararse culminaría con la aparición del carcinoma
Otro de los posibles mecanismos de carcinogénesis observado en sujetos infectados por H. pylori es el aumento del grado de proliferación celular, condicionado por un incremento en la producción del factor de crecimiento epidérmico (TGF-b) con relación a factores liberados por el germen y a la situación de hipergastrinemia secundaria, que remite tras la erradicación bacteriana. El aumento en la velocidad de replicación celular podría aumentar la posibilidad de mutaciones espontáneas, algunas de las cuales podrían incorporarse de forma permanente al ciclo celular, y de esta forma perpetuar un nuevo linaje celular completamente diferente al que codificaría el DNA del huésped en condiciones normales. La ureasa liberada por el bacilo para su protección, causa efectos similares al TGF-b, esta enzima genera grandes cantidades de amonio para la neutralización del pH en su periplasma, y aunque promueve su protección, es otra de las sustancias que se ha implicado en la acción estimuladora de la replicación celular produciendo asimismo un efecto celular mutagénico.(27)
FIGURA 7: PATOGENIA DEL CÁNCER GÁSTRICO
|