Genética Enfermedad de Fabry: Desde el Aminoácido a la Clínica
Genética de la Enfermedad de Fabry
Las células hepáticas humanas, sintetizan α-Gal-A
como un precursor de peso molecular 58000 kD y es posteriormente procesado
a una forma de peso molecular de 49000 kD. el ADN de la enzima humanaha sido clonado y codifica la secuencia
proteica completa de la enzima madura [11]. En 1996, Miyamura y col.[7] ubicaron en el brazo largo del
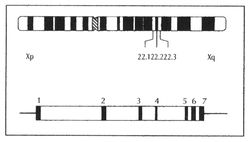
cromosoma X (Xq22.1) (Figura 4),la secuencia genómica de 14Kb y en el
2002, Pastores y col. [8] encontraron que el cromosoma contiene 7
exones que codifican el polipéptido α-Gal-A de 429 aminoácidos, incluyendo el
péptido señal de 31 residuos en el extremo amino terminal. Las diferentes mutaciones en los genes
estructurales lisosomales favorecen la heterogeneidad, que se observa en la
expresión clínica de estas enfermedades. Por ejemplo, una mutación en un gen
que codifica para una enzima lisosomal puede causar pérdida total de la
actividad enzimática, mientras que otras mutaciones en el mismo gen, puede
resultar en sólo una alteración parcial de la actividad enzimática, por
consiguiente el curso clínico de la enfermedad sería menos severo. Otras
mutaciones generan modificaciones post-translacionales o la afinidad a
diferentes sustratos [4].
Fig. 4.- En la parte
superior de la figura se puede apreciar la localización en el cromosoma X del
gen de la α-Gal-A. En la parte inferior se representa un esquema del gen de la
α-Gal-A. En negro destacan los exones y en blanco los intrones (Tomado de Torra
R y Ballarín J, 2003)[12]
Aproximadamente el 57% de los alelos
enfermos son mutaciones en donde un solo aminoácido es sustituido por otro
(mutación missense), 11% sin sentido (mutación non sense), 18% supresiones
genéticas parciales, 6% inserciones y 6% defectos en el procesamiento del RNA
debido a uniones genéticas aberrantes. Se han encontrado mutaciones en los 7
exones. En la enfermedad de Fabry muchas de estas mutaciones son sustraídas y
confinadas a una sola familia. El defecto en el gen α-GAL A, podría
estar asociado en varios niveles, que estarían bien sea a un nivel
de la proteína como tal o en la actividad enzimática indetectable (como
resultado de una transcripción inestable de RNA), o con niveles perceptibles de
la proteína enzimática, pero con actividad enzimática indetectable (mutaciones
que afectan el sitio catalítico) y/o actividad de residuos de α-GAL A
que se pueda medir (mutaciones que afectan el Holding proteico, la unión a
sustrato o la tasa de recambio) [8]. La monosomía del cromosoma X se produce
por una compensación de dosis, un sistema de todos los organismos con
determinación sexual XY, la cual trae equilibrio de la expresión de muchos
genes ligados al cromosoma X en mujeres y hombres. En mamíferos, este sistema de
compensación incluye el ?silenciamiento? de los genes en el cromosoma X el cual
se conoce como Inactivación Cromosómica. En 1961 Lyon MF [12], planteó
que en el desarrollo temprano, los cromosomas X en el embrión femenino, están
inactivados al azar, por ello en algunas células de la madre, el cromosoma X se
consideraba inactivado. Una vez que ocurría la inactivación en una célula, el
patrón de inactivación del cromosoma X se transformaba en una parte fija de la
herencia somática de las células. Por consiguiente, en las hembras se observaba
como un mosaico, con algunas células expresando genes del cromosoma paterno y
otras expresando genes del cromosoma materno [13]. En ratones, los
dos cromosomas X del embrión femenino temprano no están diferenciados, tanto
citológica como funcionalmente; ambos cromosomas X son activos. La inactivación
en la línea germinativa es cíclica. En las hembras, un cromosoma X es
inactivado durante la fase mitótica u oogonial, mientras que ambos cromosomas X
son activados en la oogénesis. En los machos, debido a que tienen solo
cromosoma X, no se presenta la inactivación en las células somáticas. La
inactivación del cromosoma X en las células somáticas generalmente es al azar,
teniendo la misma probabilidad de inactivación tanto con los cromosomas X
materno como paterno [13].
El proceso de inactivación de los
cromosomas se divide en tres fases:1.- Iniciación; 2.- Extensión, y 3.-
Mantenimiento.
1.- Iniciación
A partir de estudios de translocación y
supresión del cromosoma X autonómico, surge la evidencia de la existencia y la
localización de un centro de inactivación del cromosoma X (XIC), el cual
interfiere con la manifestación de la inactivación del mismo. Un gen único que
mapea para XIC, tiene propiedades particulares, sugiriendo que puede ser
el locus esencial para el inicio de la inactivación del cromosoma X, y el mismo
ha sido designado como Transcriptor Específico de la Inactivación de X
(XIST). Esta es una fase heterocromática, de lenta replicación, con
hipermetilación de las regiones promotoras resistente a nucleasa. La metilación
de la citosina ocurre enzimaticamente después de la síntesis de DNA, y en
mamíferos está restringida al dinucleótido 5´-CpG-3`. Esta mutación
reprime la transcripción, y en el genoma en células normales, la gran mayoría
de ellos se encuentran en el cromosoma X inactivo. Esto es importante en el
mantenimiento del estado de represión de los genes ligados al cromosoma X
inactivo, e incluso juega un papel importante en el establecimiento de la inactivación
del cromosoma X [13]. Además, en esta fase ocurre una
hipoacetilación (relacionada con el silencio genético) en los residuos de
lisina en las histonas, enriquecida para la Histona Macro H2A; y
muy importante, cubierta por XIST-RNA. Si el extremo 5? del gen XIST es suprimido, el
cromosoma no puede ser inactivado, mientras que la supresión de 3? del gen, asegura la
inactivación. Se ha demostrado que a pesar de que XIST se inicie en 5?, se transcribe un
transcriptor antisentido, llamado TSIX, desde el promotor 3? al locus XIST. Al igual que
XIST, TSIX produce un RNA nuclear no traducido, y juega un papel importante en
la protección del cromosoma de ser cubierto por XIST e inactivado (regulación
de XIST). En las etapas embrionarias muy tempranas, XIST sólo se transcribe en
el embrión femenino, y sólo del cromosoma X paterno. Por otro lado, TSIX se
transcribe tanto en embriones masculinos o femeninos, y sólo del cromosoma X
materno. En las células somáticas, la mutilación está incluida en el mantenimiento
de un gen XIST silente en el cromosoma X activo; pero en el embrión temprano,
la evidencia disponible indica que la mutilación no está comprometida con la
expresión de XIST. Al mismo tiempo, en el que se está formando XIST-RNA en el
embrión temprano, la histona macro H2A, ha mostrado asociarse preferencialmente
con la inactivación del cromosoma X.
2.- Extensión.
La extensión de la inactivación es un
enigma. Se ha planteado que la extensión de la inactivación ocurre de una
manera dominio por dominio, tal vez mediada por un locus de control
llamado Región de Inactivación unida al cromosoma X.
3.- Mantenimiento.
La mutilación y la replicación tardía son
factores críticos para el mantenimiento del silenciamiento del cromosoma X
inactivo [13]. En los estados de mórula tardía o
blastocisto temprano, se observan evidencias tempranas del XCI: asincronía de
la replicación del DNA entre los cromosomas, expresión génica diferencial y
formación de la cromatina sexual (cromosoma X inactivo). En estas células, el
cromosoma X paterno es inactivado preferencialmente. En el momento de la
gastrulación ocurre la inactivación al azar del cromosoma X; una vez que ocurre
la inactivación, toda la descendencia de una célula puede tener el mismo
cromosoma X silente [13]. En ambos sexos, los genes responsables de
las enfermedades ligadas al cromosoma X tienen riesgos clínicos diferentes.
Debido a que las mujeres tienen 2 cromosomas X, deben ser heterocigoto u
homocigoto para un gen mutante, y el gen alelo mutante puede demostrar
expresión recesiva o dominante. En las mujeres, con frecuencia la expresión
genética es variable e influenciada por la inactivación al azar del cromosoma
X. Por otra parte, los hombres tienen solamente 1 cromosoma X, así que ellos
son más propensos a mostrar un fenotipo completo. Por consiguiente, los
términos ?dominante ligado al cromosoma X, ?recesivo ligado al cromosoma X?
hacen referencia solamente a la expresión de las mutaciones en mujeres. En
general, las enfermedades recesivas son muy raras, debido a que la capacidad
biológica reducida de los homocigotos sirve para remover el gen mutante de la
población [4]. Muchas de las deficiencias enzimáticas en
las enfermedades recesivas, involucran enzimas de las vías catabólicas,
normalmente las enzimas que degradan moléculas orgánicas de la dieta diaria,
como son la galactosa, la fenilalanina y el ácido titánico (Síndrome de
Refsum). Cuando la deficiencia de la enzima afecta una hidrolasa ácida
(desórdenes de depósito lisosomal), el sustrato el cual usualmente es un lípido
complejo o un polisacárido, el mismo se acumula dentro de los lisosomas
edematizados. En las mujeres, la inactivación del
cromosoma X al azar es el determinante más importante de la expresión de los
desórdenes ligados al cromosoma X. Muchos individuos son asintomáticos, otros
presenta síntomas moderados y otros tienen manifestaciones severas. La
frecuencia de las alteraciones fenotípicas detectables, depende de un examen
cuidadoso de los heterocigotos y de la edad en que se evalúen los mismos. La
enfermedad de Fabry, junto con la Hemofilia A, la Distrofia Muscular
de Duchenne, son ejemplos de enfermedades en las que pueda observarse expresión
clínica en mujeres [4]. El análisis del gen de α-Gal-A, en
la enfermedad de Fabry, mostró la existencia de lesiones moleculares
heterogéneas, tales como mutaciones puntuales y reordenamiento genético
parcial. Toshio y col (1999)[14] utilizando en ratones el modelo
experimental de la enfermedad de Fabry, señalaron que los genes que codifican
para α-Gal-A , son muy similares en tamaño, organización y secuencia
nucléotidica de las regiones de codificación tanto en humanos como en ratones.
Se han reportado más de 70 mutaciones en el gen que codifica para esta
enzima, muchas de las cuales resultan en el fenotipo clásico de falta de
actividad de la α-Gal-A[7]. Entre alguna de estas
mutaciones se ha descrito unacon diferentes codones de detención
prematura, y todos los pacientes hemizigotos con dichas mutaciones, incluyendo
E398X, manifiestan el fenotipo clásico de la enfermedad y probablemente solo
fallan en 32 aminoácidos del extremo carboxilo terminal. Por otra parte se ha
descrito que 26 de 28 residuos de aminoácidos deben ser eliminados
proteolíticamente del extremo carboxilo terminal de la enzima
galactosidasa, para generar el péptido final. Esto sugiere que la región
carboxilo terminal debe ser uno de los extremos activos o debe tener influencia
en la conformación del sitio activo [7]. Tanto clínica como bioquímicamente, la
enfermedad de Fabry muestra una alta variabilidad fenotípica. Se podría
observar heterogeneidad intrafamiliar, lo que se explica en pacientes con la
misma mutación, por la presencia de genes modificados que cooperan en la
expresión de α-Gal-A. Las mutaciones más frecuentes, descritas en la
enfermedad de Fabry son las lesiones genéticas R227Q (con compromiso
neurológico) y R220X, y se correlacionan con la forma clásica de la enfermedad.
En el 2003, Morrone y col. [15] explicaron que su alta frecuencia en
las mutaciones se debe a la presencia de CpG en el gen de α-Gal-A. El
93% de los genotipos está asociado con la enfermedad clásica, mediante
mutaciones sin sentido y con una lectura de los codones en manera diferente,
las cuales causan una terminación prematura de la síntesis proteica. Muchas
mutaciones en donde un aminoácido es sustituido por otro, afectan los sitios
catalíticos, sitios de dimerización o la forma intricada tridimensional de las
proteínas [8]. Morrone y col, (2003) tambiénobservaron
que las mutaciones que más se relacionan con daño renal son las S78X, C126-127,
CATG y la A352D. Se
observó en los leucocitos periféricos la presencia de α-Gal-A residual
cuantificable, asociada con un manifestación tardía de la insuficiencia renal
crónica, con un menor contenido de ceramida trihexosa y menor grado de daño
histológico renal. Además se ha observado que las mutaciones conservativas
donde un aminoácido es sustituido por otro estaban asociadas con una mayor
sobrevida renal, comparada con las mutaciones no conservativas [8,15]. Sin embargo, se ha observado que en
hombres algunas de esas mutaciones con sustituciones únicas de aminoácidos,
localizadas en la región carboxilo terminal llevan a variantes atípicas de la
enfermedad de Fabry, con manifestaciones limitadas al corazón. Se ha sugerido
que esta variedad debe ser más común de lo que se piensa, dado que se presenta
en pacientes masculinos con hipertrofia ventricular izquierda inexplicable [7].
Solo se han reportado 18 genotipos asociados con la variante cardiaca de la
enfermedad de Fabry; y la mayoría de ellas son mutaciones en donde está
involucrado un solo aminoácido. Muchas de esas mutaciones producen
inestabilidad de la proteína, debido a un empaquetamiento inapropiado de la
estructura proteica, y una alteración en el sitio de glicosilación (N215S). Han
sido reportadas cuatro mutaciones (R112H, R301Q, G328R y R404) asociadas a la
variante cardíaca en una familia y asociadas a Fabry clásico en otra [7]. No están completamente claras, las bases
para las variaciones fenotípicas de la enfermedad de Fabry, aunque puede
atribuirse en parte a la heterogeneidad de las mutaciones causales e incluso al
tipo de sangre del paciente. Los pacientes con grupo sanguíneo B y AB (con
residuos terminales de α- galactosil en sus membranas celulares) pueden tener
un mayor sustrato corporal y tienen una enfermedad más agresiva comparada con
pacientes con grupos sanguíneo A u O. Esta hipótesis no ha sido
sistemáticamente estudiada, y se necesita más información de la historia
natural de la patología que permita conocer los diversos factores que influyen
en la expresión de la enfermedad de Fabry [8].
NOTA:Toda la información que se brinda en este artículo es de carácter investigativo y con fines académicos y de actualización para estudiantes y profesionales de la salud. En ningún caso es de carácter general ni sustituye el asesoramiento de un médico. Ante cualquier duda que pueda tener sobre su estado de salud, consulte con su médico o especialista.
Instituto de Medicina Tropical - Facultad de Medicina - Universidad Central de Venezuela.
Elaborado por el Centro de Análisis de Imágenes Biomédicas Computarizadas CAIBCO, caibco@ucv.ve
Este portal ha sido desarrollado gracias al apoyo del Fonacit