Biología celular
El receptor del factor de crecimiento epidérmico
Regulación del EGFR
Una vez que las diversas señales han sido transmitidas por el EGFR activado, actúan diversos mecanismos capaces de inactivar al receptor con objeto de evitar la estimulación mitogénica continuada de la célula. Así, existen sistemas rápidos y lentos de inactivación del receptor. Los primeros pueden actuar en minutos, tales como fosforilaciones del receptor por proteínas quinasas reguladoras o la interacción con el complejo Ca2+/calmodulina; y los últimos actúan en horas, como es la internalización del receptor y su degradación proteolítica.
Entre las proteínas quinasas capaces de regular al EGFR inhibiéndolo, citaremos a la PKC (Hunter et al., 1984), la proteína quinasa II dependiente de calmodulina (CaMPK-II, por calmodulin-dependent protein kinase II) (Countaway et al., 1990), la proteína quinasa activada por mitógenos (MAPK, por mitogen-activated protein kinase) (Northwood et al., 1991; Takishima et al., 1991), la proteína quinasa dependiente de AMPc (PKA, por protein kinase A) (Barbier et al., 1999) y la proteína quinasa dependiente de ciclina p34cdc2 (Kuppuswamy et al., 1993). Por otro lado, distintas fosfoproteína fosfatasas, como las fosfatasas 1 y 2A, parecen estar implicadas en la defosforilación del EGFR, puesto que cuando éstas son inhibidas se produce una hiperfosforilación del mismo (Hernández-Sotomayor et al., 1991). Más recientemente, se ha demostrado que la tirosina quinasa TCPTP (por T-cell protein-tyrosine kinase) está implicada en la desfosforilación de los residuos de fosfotirosina del EGFR y la posterior disminución de la señalización mediada por éste (Tiganis et al., 1999).
Tras la activación del EGFR por sus ligandos, uno de los primeros eventos observados en diversos tipos celulares es el aumento transitorio de la concentración del Ca2+ libre citosólico, proceso que está mediado por la PLCg y la PLA2, como hemos visto anteriormente (Villalobo et al., 2000). Dentro de los mecanismos de desactivación del EGFR, describiremos con más detalle los procesos dependientes de la señal del calcio generada por el receptor, como son las fosforilaciones mediadas por la PKC y la CaMPK-II; y la acción directa del complejo Ca2+/calmodulina sobre el receptor.
La PKC presenta al menos 12 isoformas diferentes que se expresan de forma desigual en los diversos tejidos, están localizadas en diferentes compartimentos intracelulares y poseen modos de acción y especificidad de sustrato diferentes (Hug and Sarre, 1993; Dekker and Parker, 1994). Así, el aumento de la concentración del Ca2+ citosólico activa a diversas isoformas de la PKC que fosforilan al EGFR en su Thr654 localizada en la región citosólica yuxtamembranal, inhibiendo su actividad tirosina quinasa y la internalización del mismo (Hunter et al., 1984; Davis, 1988; Lund et al., 1990; Countaway et al., 1990; Morrison et al., 1996) (ver Fig. 8). Sin embargo, la fosforilación de la Thr654 no parece ser responsable de la pérdida de la afinidad del receptor por el EGF (Davis, 1988). Por otro lado, se ha sugerido que la baja estequiometría de fosforilación de la Thr654 podría indicar que ésta no es suficiente para mediar la inhibición del receptor por la PKC (Friedman et al., 1989; Countaway et al., 1990). Se ha propuesto que el efecto inhibidor de la PKC podría además estar mediado por la MAPK, que es capaz de fosforilar al receptor en su Thr669 inhibiéndolo (Northwood et al., 1991; Takishima et al., 1991; Morrison et al., 1993, 1996). Para estudiar estos efectos se han generado EGFR mutados en los que se han sustituido la Thr654 y la Thr669 por sendos restos de ácido glutámico, con objeto de que la carga negativa de éste mimetice los efectos del fosfato de estos restos fosforilados. Estos estudios han demostrado que la introducción de una carga negativa en la región citosólica yuxtamembranal del receptor no parece ser suficiente para producir la inhibición del mismo (Morrison et al., 1993). No obstante, aunque en diferentes líneas celulares se ha observado que la PKC es capaz de inhibir al EGFR por un mecanismo dependiente de la vía de la MAPK, esta inhibición no parece ser debida solamente a la fosforilación del receptor mediada por la PKC o por la MAPK, ya que existen evidencias de que la PKC podría activar, a través de la MAPK, a una proteína fosfatasa capaz de desfosforilar al receptor produciendo la inactivación del mismo (Morrison et al., 1996) (ver Fig. 8).
La CaMPK-II también interviene muy significativamente en la inhibición del EGFR mediada por el incremento de la concentración del Ca2+ libre citosólico. Esta quinasa es capaz de fosforilar al EGFR en sus residuos Ser1046 y Ser1047, lo que induce la inhibición de la actividad tirosina quinasa del receptor y su internalización (Countaway et al., 1992; Theroux et al., 1992). Adicionalmente, se han descrito otros sitios de fosforilación del receptor por la CaMPK-II como son la Ser744, la Ser1142 y la Ser1057, lo cual produce también la inhibición de la actividad tirosina quinasa del mismo (Feinmesser et al., 1999) (ver Fig. 8). Por otro lado, el receptor ErbB2/Neu también está regulado por esta quinasa (Feinmesser et al., 1999). Los sitios de fosforilación de estos receptores por la CaMPK-II poseen funciones reguladoras muy importantes, ya que las células que expresan receptores mutados que carecen de estos sitios de fosforilación se transforman incrementando su potencial oncogénico (Countaway et al., 1992; Feinmesser et al., 1999).
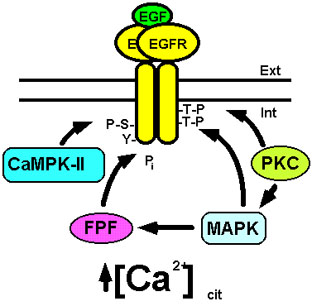
Figura 8. Retroinhibición del EGFR por la señal del Ca2+. El aumento transitorio de la concentración de Ca2+ citosólico da lugar a la activación de la PKC y la CaMPK-II que fosforilan al EGFR en residuos de treonina (-T-P) y serina (-S-P), respectivamente. La MAPK también fosforila residuos de treonina en el receptor. Estas fosforilaciones inhiben la actividad tirosina quinasa del mismo. Adicionalmente, la MAPK activa, mediante una vía dependiente de PKC, a una fosfoproteína fosfatasa (FPF) que desfosforila los restos de tirosina (Y) fosforilados del EGFR desactivándolo. Para más detalles ver texto.

La señal del Ca2+ generada por el EGFR produce además la formación del complejo Ca2+/calmodulina. Este complejo ejerce una acción reguladora directa sobre el EGFR, independientemente de la acción mediada por la CaMPK-II, que también actua sobre el receptor como hemos visto anteriormente. Así, nuestro grupo ha demostrado que el complejo Ca2+/calmodulina es capaz de unirse directamente al EGFR e inhibir su actividad tirosina quinasa (San José et al., 1992; Benguría and Villalobo, 1993; Benguría et al., 1995; Villalobo et al., 2000). Este proceso parece ocurrir por la interacción del complejo Ca2+/calmodulina con un sitio de unión de la misma localizado en la región citosólica yuxtamembranal del receptor que está muy conservado filogenéticamente desde las aves hasta el hombre (San José et al., 1992; Martín-Nieto and Villalobo, 1998). En este sitio se encuentra la Thr654, que, como hemos visto anteriormente, es fosforilada por la PKC, siendo la unión de la calmodulina y la fosforilación de este residuo procesos mutuamente excluyentes (Martín-Nieto and Villalobo, 1998; Villalobo et al., 2000) (ver Fig. 9). Aunque ambas señales inducen la inhibición de la actividad tirosina quinasa del receptor, estos procesos podrían tener significados fisiológicos diferentes. Por un lado, la fosforilación del EGFR por la PKC parece constituir una señal de inhibición de la internalización del receptor mediada por la unión del ligando (Lund et al., 1990). Por el contrario, nosotros hemos propuesto que la unión del complejo Ca2+/calmodulina al receptor podría constituir una señal de internalización de éste (Martín-Nieto and Villalobo, 1998; Villalobo et al., 2000).
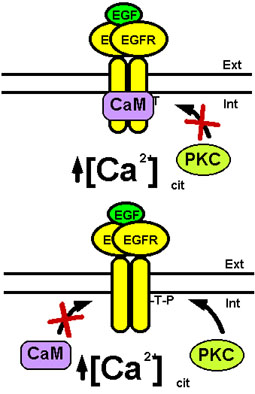
Figura 9. Regulación mutuamente excluyente del EGFR por la calmodulina y la PKC. (Panel superior) El incremento de la concentración de Ca2+ citosólico induce la formación del complejo Ca2+/calmodulina (CaM) que se une al EGFR inhibiendo su actividad tirosina quinasa. Este proceso previene la fosforilación del receptor por la PKC. (Panel inferior) La fosforilación del EGFR por la PKC previene la unión del complejo Ca2+/calmodulina al receptor. Para más detalles ver texto.
La relación entre la calmodulina y el EGFR parece ser más compleja de lo explicado hasta ahora, ya que también hemos demostrado que este receptor es capaz de fosforilar a la calmodulina en su Tyr99 con una estequiometría cercana a 1 (mol/mol), en ausencia de Ca2+ y en presencia de un cofactor básico (San José et al., 1992; Benguría and Villalobo, 1993; Benguría et al., 1994, 1995; De Frutos et al., 1997; Benaim et al., 1998; Palomo-Jiménez et al., 1999; Villalobo et al., 2000). Esta fosforilación podría constituir un sistema de regulación de la calmodulina, modulando así distintos sistemas enzimáticos o estructurales dependientes de ésta. En este contexto, nuestro grupo también ha demostrado que la calmodulina fosforilada por el EGFR, y libre de calmodulina no fosforilada, pierde la capacidad de activar a la fosfodiesterasa de nucleótidos cíclicos de corazón bovino (Palomo-Jiménez et al., 1999) (ver Fig. 10). Sin embargo, parece ser que esta regulación no es universal, ya que otros autores han demostrado que preparaciones similares de fosfo(Tyr)calmodulina activan a la fosfodiesterasa de nucleótidos cíclicos de cerebro bovino como lo hace la calmodulina no fosforilada (Corti et al., 1999). Esta aparente discrepancia puede ser debida a que en el corazón y en el cerebro bovino existan diferentes isoformas de esta enzima, o a pequeñas diferencias en el estado de fosforilación de la calmodulina (Villalobo et al., 2000). Por otro lado, la fosfo(Tyr)calmodulina parece ejercer una acción potenciadora de la actividad tirosina quinasa del EGFR en presencia, pero no en ausencia, de EGF. Esto ha sido propuesto en base a experimentos en los que se permitió en una primera fase la acumulación de calmodulina fosforilada por el receptor, y posteriormente se ensayó en una segunda fase la fosforilación de otro sustrato del mismo, observándose un pronunciado incremento de la fosforilación del segundo sustrato cuando se comparaba con controles en los que no se había acumulado calmodulina fosforilada (Villalobo et al., 2000) (ver Fig. 10).
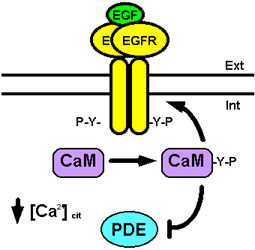
Figura 10. Fosforilación de la calmodulina por el EGFR y mecanismos reguladores. Cuando la concentración de Ca2+ citosólico es baja, al comienzo del proceso de estimulación mitogénica, el EGFR fosforila a la calmodulina (CaM) generándose de esta forma fosfo(Tyr)calmodulina (CaM-Y-P). Ésta por un lado estimula más aún la actividad tirosina quinasa del EGFR, y por otro pierde la capacidad de estimular a ciertas fosfodiesterasas de nucleótidos cíclicos (PDE). Para más detalles ver texto.
Con objeto de documentar la acción reguladora sobre el receptor de un sistema de naturaleza diferente al de los mencionados hasta ahora, indicaremos que el EGFR parece estar también bajo el control regulador del óxido nítrico, agente gaseoso fisiológico de vida efímera que es sintetizado a partir de L-arginina por la óxido nítrico sintetasa, enzima que también es regulada por el complejo Ca2+/calmodulina. El óxido nítrico regula múltiples funciones celulares y sistémicas incluida la proliferación celular. Así, hemos puesto en evidencia que dicho gas es capaz de reaccionar covalentemente con grupos -SH del receptor y que dicho proceso (denominado S-nitrosilación) da lugar, de una forma reversible, a la inhibición de la transfosforilación del receptor y de la fosforilación de sustratos exógenos, lo que podría contribuir a la inhibición de la proliferación celular inducida por EGF en condiciones de estrés nitrosativo (Estrada et al., 1997) (ver Fig. 11).
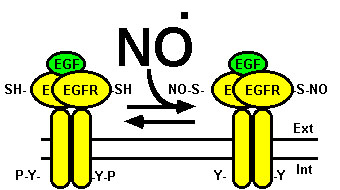
Figura 11. Regulación del EGFR por óxido nítrico. El óxido nítrico (NO·) S-nitrosila al EGFR inhibiendo su actividad tirosina quinasa. Proceso que es revertido por agentes reductores. La localización de los grupos -SH susceptibles de ser S-nitrosilados se desconoce. En este esquema se representan arbitrariamente en el dominio extracelular. Para más detalles ver texto. |